Dieser Themenbereich erklärt zum einen grundlegende Begriffe und Definitionen. Zum anderen werden onkologische Behandlungsstrategien, Methoden und Mechanismen gezeigt. Medikamentöse Maßnahmen werden in historischem Kontext über die aktuellen Ansätze der Gegenwart bis hin zu sich abzeichnenden, kommenden Entwicklungen geschildert. Da die onkologische Therapie ein polypharmazeutischer Therapieansatz ist, sind Wechselwirkungen verschiedener Wirkstoffe sehr wahrscheinlich. Daher wird auf spezifische Wechselwirkungen und den professionellen Umgang damit ebenfalls eingegangen.
Grundlagen
Norbert Schleucher, Jürgen Barth
1 Onkologische Therapieziele
Einer onkologischen Therapie können verschiedene Zielsetzungen zugrunde liegen, die vor Therapiebeginn definiert werden müssen.
1.1 Kurative Therapie
Eine kurative Therapie hat das Ziel, eine Heilung im Sinne einer dauerhaften Tumorfreiheit zu erreichen. Voraussetzung dafür ist bei einem operativen Vorgehen die vollständige Entfernung des Tumors mit einem ausreichenden Sicherheitsabstand (R0-Resektion). Voraussetzung bei einer medikamentösen oder radioonkologischen Therapie ist eine vollständige Tumorrückbildung (siehe auch Abschnitt Remissionsdefinitionen).
1.2 Adjuvante Therapie
Das Ziel einer adjuvanten Therapie ist die Reduktion des Rezidivrisikos durch Elimination bildgebend nicht darstellbarer Mikrometastasen. Dabei erfolgt eine medikamentös-zytostatische Behandlung, eine Strahlentherapie oder eine Kombination beider Verfahren im Anschluss an eine chirurgisch komplette Tumorresektion. Eine adjuvante Therapie wird in bildgebend und laborchemisch (= negative Tumormarker) vollständiger Tumorfreiheit durchgeführt. Sie ist erforderlich bei Tumoren, die ein hohes postoperatives Rezidivrisiko aufweisen, wie beispielsweise das Mammakarzinom, das kleinzellige Bronchialkarzinom oder das lokal fortgeschrittene Kolonkarzinom.
1.3 Erhaltungstherapie
Unter einer Erhaltungstherapie versteht man eine medikamentöse Therapie, die beginnt nachdem der Patient auf die vorangehende Behandlung in Form einer kompletten oder partiellen Remission (CR/PR) oder mit Erreichen einer stabilen Erkrankung (SD) angesprochen hat. Das Ziel einer Erhaltungstherapie ist die Verlängerung des progressionsfreien Überlebens, d.h. die Verlängerung der Zeit bis zur nächsten Therapielinie. Damit wird eine Erhaltungstherapie in der Regel bis zur Erkrankungsprogression durchgeführt (sofern sie ausreichend verträglich ist und keine gravierende Toxizität auftritt). Oftmals resultiert daraus auch eine Verlängerung des Gesamtüberlebens.
Ein historisches Beispiel einer Erhaltungstherapie ist die Interfon-Erhaltung bei malignen Lymphomen. Diese wurde durch die Erhaltungstherapie mit Rituximab verdrängt, welche auch heute noch bei verschiedenen Lymphomen (follikuläres NHL, Mantelzell-Lymphom) therapeutischer Standard ist. Beim multiplen Myelom ist die Erhaltungstherapie mit Lenalidomid ebenfalls ein therapeutischer Standard, auch nach Durchführung einer Stammzelltransplantation. Auch eine Erhaltung mit Bortezomib ist beim Myelom möglich.
Auch bei soliden Tumoren sind Erhaltungstherapien weit verbreitet. Vorreiter war hier das nicht-kleinzellige Bronchialkarzinom, bei dem Erhaltungstherapien mit Pemetrexed (Paramount-Studie) oder Erlotinib (Saturn-Studie) bereits vor etwa 10 Jahren in die Praxis eingeführt wurden. Heute erfolgen diese Erhaltungsbehandlungen bei NSCLC jedoch auf Basis immunologischer Therapien mit Checkpoint-Inhibitoren: Die Therapie beginnt mit einer Chemo-Immuntherapiekombination und nach 2-4 kombinierten Behandlungszyklen wird die Chemotherapie beendet und die Immuntherapie bis zum Progress fortgeführt. Beispiele sind die Durvalumab-Erhaltung im Stadium III B nach kombinierter Chemo-Strahlentherapie, wobei in diesem Setting zuerst die Chemo-Radiotherapie erfolgt und im Anschluss die Durvalumab-Erhaltung, oder die Keynote 189 Studie, bei der mit kombinierter Chemo-Immuntherapie gestartet wird und nach 4 Zyklen auf die Pemetrexed-Erhaltung deeskaliert wird. Beim nicht-kleinzelligen Bronchialkarzinom sind auch Erhaltungstherapien mit Atezolizumab in Kombination mit Bevacizumab (VEGF Antikörper) möglich (IMPower 150 Studie) oder Erhaltungstherapien mit zwei Checkpoint-Inhibitoren Nivolumab plus Ipilimumab in Kombination (Checkmate 9LA Studie). Bei der letztgenannten Kombination erfolgen nur noch 2 Behandlungszyklen mit platinbasierter Chemotherapiedoublette und den 2 Checkpoint-Inhibitoren, danach wird die Behandlung mit der dualen Checkpoint-Inhibitor-Kombination fortgeführt.
Beim kolorektalen Karzinom erfolgen Erhaltungstherapien in Form von Deeskalationen der Therapie. Die Kombinationstherapie FOLFOX, FOLFIRI oder FOLFIRNOX in Kombination mit einem EGFR- oder VEGF-Antikörper wird im Verlauf auf 5-FU/Folinsäure oder Capecitabin plus Antikörper deseskaliert und dann bis zum Progress fortgeführt.
Ein weiteres Beispiel für eine Erhaltungstherapie ist die Avelumab-Erhaltung beim Harnblasenkarzinom (Javelin Bladder 100 Studie) nach Ansprechen auf eine Platin / Gemcitabintherapie.
Beim Ovarialkarzinom werden in der ersten und zweiten Therapielinie Erhaltungstherapien mit PARP-Inhibitoren durchgeführt, in der ersten Linie auch in Kombination mit Bevacizumab.
Formal kann auch die antihormonelle Therapie beim Mammakarzinom nach Erreichen einer Remission durch eine Chemotherapie als Erhaltungstherapie aufgefasst werden (antihormonelle Erhaltung). Im Gegensatz dazu ist die kombinierte Therapie mit einem Aromatasehemmer plus CDK 4/6 Inhibitor, die über den gesamten Therapieverlauf nicht modifiziert wird KEINE Erhaltungstherapie sondern eine Therapie bis zum Progress.
1.4 Neoadjuvante (= präoperative) Therapie
Eine neoadjuvante Therapie hat das Ziel, die zum Diagnosezeitpunkt bestehende Tumorlast zu minimieren und dadurch die Resultate der chirurgischen Tumorresektion zu verbessern bzw. einen fortgeschrittenen, inoperablen Tumor sekundär resektabel zu machen. Es kommen die zytostatische Chemotherapie, die Bestrahlung oder die kombinierte Chemostrahlentherapie zum Einsatz. Eine neoadjuvante Behandlung erfolgt bei fortgeschrittenen Tumoren, die technisch nicht resektabel sind, mit dem Ziel, diese Tumoren sekundär resezieren zu können. Weiter erfolgen neoadjuvante Chemotherapien bei Tumoren, die zwar technisch, nicht aber prognostisch resektabel sind, d. h. bei fortgeschrittenen Tumoren mit schlechter Prognose trotz kompletter Resektion. Eine neoadjuvante Behandlung sollte immer dann angestrebt werden, wenn adjuvante Therapiemodalitäten keine Prognoseverbesserung erreichen können. Als Beispiele gelten das nicht-kleinzellige Bronchialkarzinom oder die Malignome von Ösophagus und Magen.
Die präoperative Reduktion der Tumorlast wird auch als Down-Staging bezeichnet, d. h. durch die neoadjuvante Therapie wird der Tumor in ein niedrigeres Stadium überführt.
1.5 Additive (= ergänzende) Therapie
Unter einer additiven Therapie versteht man alle medikamentösen oder radiotherapeutischen Maßnahmen nach einer inkompletten chirurgischen Resektion, d. h. bei Verbleib mikroskopischer (R1-Resektion) oder makroskopischer (R2-Resektion) Tumorreste nach einer Operation.
1.6 Palliative Therapie (palliative Chemo-/Strahlentherapie)
Das Ziel einer palliativen Therapie ist die Verlängerung der Überlebenszeit bei nicht heilbaren Patienten sowie die Linderung tumorbedingter Symptome bzw. der Erhalt der Lebensqualität. Die Basis der palliativen Behandlung bildet zumeist die Chemotherapie.
Bei einer palliativen Chemotherapie ist die Therapieintensität zur Vermeidung von Nebenwirkungen geringer als bei adjuvanten oder neoadjuvanten kurativen Therapien. Bei palliativen Chemotherapien ist es von besonderer Bedeutung, die erwartete Wirkung in Relation zu den möglichen therapieassoziierten Nebenwirkungen zu setzen.
Es können jedoch auch Strahlentherapie und Chirurgie in ein palliatives Konzept integriert werden, z. B. palliative Bestrahlung einer schmerzhaften Knochenmetastase, Anlage einer Gastroenterostomie bei stenosierenden Darmtumoren oder operative Fixation frakturgefährdeter Wirbelkörper.
Neben diesen „spezifischen“ Therapien gehören beispielsweise auch Schmerz- und Ernährungstherapien in den Bereich der palliativen Therapie. Die begriffliche Abgrenzung zu supportiven und symptomatischen Therapien ist fließend.
1.7 Supportive (= unterstützende) Therapie
Als supportive Maßnahmen werden alle unspezifischen Maßnahmen – neben onkologisch spezifischer Chemotherapie, Strahlentherapie und Operation – zusammengefasst. Darunter fallen z. B. Begleitmaßnahmen gegen die Nebenwirkungen einer Chemo- oder Strahlentherapie, wie die Gabe von Antiemetika bei Übelkeit, Antibiotika bei Infektionen, Bluttransfusionen bei Anämie oder parenterale Ernährung bei Tumorkachexie. Die bestmögliche Supportivtherapie unter Ausschöpfung aller zur Verfügung stehenden Möglichkeiten wird als „best supportive care“ (BSC) bezeichnet.
1.8 Symptomatische Therapie
Die symptomatische Therapie ist ein Teilgebiet der Supportivtherapie. Eine symptomatische Behandlung soll tumorbedingte Beschwerden lindern, sie hat jedoch keinen Einfluss auf den Verlauf des Tumorleidens. Beispiele sind die Gabe von Schmerzmitteln, fiebersenkenden Medikamenten oder Hustenblockern.
1.9 Individualisierte Therapieansätze
Neue molekulare Therapiemöglichkeiten haben dazu geführt, die Systemtherapie für die Patienten individuell zu gestalten und auszuwählen. So kommt bei der Therapie des nicht-kleinzelligen Bronchialkarzinoms heute der histologischen Sub-Entitiät eine wichtige Bedeutung bei der Auswahl der Chemotherapie zu, z. B. kein Pemetrexed und kein Bevacizumab bei Plattenepithelkarzinomen zu applizieren. Darüber hinaus führen jedoch Überexpressionen bestimmter Wachstumsfaktorrezeptoren zu Therapiemöglichkeiten, die sich an den biologischen Gegebenheiten des Tumors orientieren. Beispiele sind die Therapie des kolorektalen Karzinoms mit dem EGFR-Antikörper Cetuximab bei EGFR-Überexpression des Tumors oder die hohen Ansprechraten von nicht-kleinzelligen Bronchialkarzinomen bei Mutation des EGF-Rezeptors. Diese vor Therapiebeginn bestimmten molekularen Marker eines Tumors werden auch als Biomarker bezeichnet. Teilweise kann anhand solcher Biomarker auch die Wirkungslosigkeit einer entsprechenden Therapie vorausgesagt werden, z. B. kras-Mutation und fehlendes Ansprechen auf Cetuximab beim kolorektalen Karzinom oder fehlendes Ansprechen auf Erlotinib beim nicht-kleinzelligen Bronchialkarzinom. Für weitere Einzelheiten siehe Abschnitt Anti-EGFR-Strategien.
1.10 Biomarker und molekular-pathologische Diagnostik
Im Zuge der zunehmenden Diversifizierung der zielgerichteten Therapien kommt der Bestimmung von Biomarkern und der molekular-pathologischen Diagnostik eine immer größere Bedeutung zu.
Dabei unterscheidet man zunächst zwischen prognostischen und prädiktiven Biomarkern.
Ein prädiktiver Biomarker hat eine Aussagekraft bezüglich des Ansprechens oder Nicht-Ansprechens einer zielgerichteten Therapie. Beispiele sind die Bestimmung der c-kit Mutation bei gastrointestinalen Stromatumoren oder die Bestimmung der EGFR-Mutation beim nicht-kleinzelligen Bronchialkarzinom, mit denen ein hervorragendes Ansprechen auf entsprechende Tyrosinkinase-Inhibitoren prätherapeutisch vorausgesagt werden kann. Auf der anderen Seite ist die Bestimmung bzw. der Nachweis einer KRAS- oder NRAS-Mutation beim kolorektalen Karzinom ein negativer Prädiktor für eine Therapie mit den EGFR-Antikörpern Cetuximab und Panitumumab. Diese sind bei Nachweis einer K- oder NRAS-Mutation nicht wirksam.
Ein prognostischer Biomarker hat eine Aussagekraft für die Prognose der zugrunde liegenden onkologischen Erkrankung, ohne dass sich daraus in jedem Fall eine Therapie ableiten lässt. Ein Beispiel war bislang die KRAS-Mutation beim nicht-kleinzelligen Bronchialkarzinom. Diese ist mit einer schlechten Prognose verknüpft und eine zugelassene zielgerichtete Therapieoption gibt es nicht (bislang galt KRAS als undrugable). Hier konnten mit Hilfe der molekularen pathologischen Diagnostik und der translationalen Forschung verschiedene KRAS-Mutationen identifiziert werden, von denen die KRAS-G12C-Mutation therapeutisch nutzbar ist. Sie ist das therapeutische Ziel des Tyrosinkinase-Inhibitors Sotorasib.
Ein Biomarker kann auch prognostisch und gleichzeitig prädiktiv sein wie beispielsweise HER2/neu beim Mammakarzinom. Vor Einführung der zielgerichteten Therapien gegen HER2/neu handelte es sich um einen prognostischen Marker, der auf eine ungünstige Erkrankungsprognose mit verkürzter Überlebenszeit hindeutete. Die HER2/neu-Überexpression gilt auch heute noch als Risikofaktor in der adjuvanten Therapie des Mammakarzinoms. Nach Einführung der zielgerichteten Therapien wie Trastuzumab, die nur bei hoher HER2/neu-Überexpression wirksam sind, handelte es sich auch um einen für die Therapie prädiktiven Biomarker und der Einsatz von Anti-HER2/neu-Therapien führt zu einer deutlichen Überlebensverlängerung.
Mit der Entwicklung von Immuncheckpoint-Inhibitoren gelang in den letzten Jahren ein Durchbruch in der Tumortherapie. Immuncheckpoint-Inhibitoren aktivieren die Immunabwehr gegen Tumoren, indem sie die immunhemmende Wirkung spezifischer, als Kontrollpunkte agierender Zelloberflächenproteine, der sogenannten Checkpoints, aufheben.
Die bisher zugelassenen Immuncheckpoint-Inhibitoren, gegen die Checkpoints CTLA 4 und PD-1/PD-L1 gerichtete monoklonale Antikörper, werden in verschiedenen Tumorentitäten wie Melanom, Lungen, Nieren, Urothelkarzinom oder Kopf-Hals-Tumoren sowie dem Hodgkin-Lymphom eingesetzt.
Wie oben bereits erwähnt dienen verschiedene Biomarker zur Personalisierung der Therapie. Für den Einsatz von verschiedenen immunonkologischen Produkten, in verschiedenen Tumorentitäten ist die Testung und das Vorhandensein des Biomarkers PD-L1 Voraussetzung für den Einsatz. Die Bestimmung und das Vorhandensein von PD-L1 als Biomarker erfüllen somit bereits viele Kriterien eines personalisierten Therapieansatzes.
Für immunonkologische Therapien gilt PD-L1 (= Programmed Death Ligand 1) somit als Biomarker für die Therapie mit Immuncheckpoint-Inhibitoren. Beim malignen Melanom konnte man die „Höhe“ der PD-L1-Expression mit dem Ansprechen auf Immuncheckpoint-Inhibitoren wie Nivolumab oder Pembrolizumab korrelieren: Bei „hoher“ Expression fand sich ein hohes und langanhaltendes Ansprechen auf die Monotherapie mit Pembrolizumab oder Nivolumab und eine kombinierte Immuncheckpoint-Hemmung gegen PD-L1 und gleichzeitig auch gegen CTLA4 mit Ipilimumab war nicht erforderlich. Bei „niedriger“ PD-L1-Expression war die therapeutische Effizienz gut, aber geringer und die Patienten haben vermehrt von einer kombinierten Therapie profitiert. Dieses Modell beim Melanom hat sich jedoch nicht uneingeschränkt auf andere Tumorentitäten übertragen lassen und insbesondere beim nicht-kleinzelligen Bronchialkarzinom ist die Datenlage heterogen. So sind Atezolizumab und Nivolumab in der Zweitlinientherapie ungeachtet des PD-L1 Status zugelassen (also auch bei negativer Expression), die Zulassung von Pembrolizumab ist jedoch an einen positiven PD-L1-Nachweis gekoppelt. In der Erstlinientherapie des nicht-kleinzelligen Bronchialkarzinoms ist eine Monotherapie mit Pembrolizumab für „hoch“ PD-L1-positive Tumoren (³ 50%) zugelassen, eine Nivolumab Monotherapie jedoch nicht.
Heterogen sind auch die molekularen Bestimmungsverfahren für PD-L1. Es existieren mindestens 4 verschiedene diagnostische Antikörper für die molekulare pathologische Diagnostik. Es können dabei sowohl Tumorzellen als auch Interstitialzellen angefärbt und analysiert werden. Die Ergebnisse werden dann in %TC oder %IC angegeben. Darüber hinaus gibt es auch Scoring-Systeme wie den Combined Positivity Score (CPS, positiv gefärbte Tumorzellen + positiv gefärbte Immunzellen geteilt durch die Gesamtzahl an Tumorzellen in Prozent) und den Tumor Proportion Score (TPS, Quotient aus positiv gefärbten Tumorzellen und Gesamtzahl der Tumorzellen in Prozent).
Neben gezielten erkrankungsspezifischen Biomarkern gibt es mit NTRK auch einen sog. tumoragnostischen Marker. Dieser ist in geringem Prozentsatz von 1–2% bei nahezu allen Tumoren positiv. Bei Positivität kann eine Behandlung mit Larotrectinib oder Entrectinib unabhängig vom zugrunde liegenden Tumor erfolgen.
Bei Erkrankungen wie dem nicht-kleinzelligen Bronchialkarzinom oder dem kolorektalen Karzinom ist es üblich, mehrere Biomarker vor Therapiebeginn zu bestimmen. Man spricht dann von einem sogenannten Biomarker-Panel. Beim kolorektalen Karzinom umfasst ein solches Panel KRAS, NRAS, BRAF und MSI. Beim nicht-kleinzelligen Bronchialkarzinom, speziell Adenokarzinom, kann ein solches Panel z.B. PD-L1, BRAF, EGFR-Mutation, ALK und ROS1 umfassen und durch NTRK, RET und c-MET zentrumsimmanent erweitert werden.
Die „Maximalvariante“ eines Biomarker-Panels ist das Next-Generation-Sequencing (NGS). Beim NGS werden nahezu alle therapierelevanten Marker einschließlich Resistenzmarkern untersucht, aber auch Biomarker, für die keine zielgerichtete Therapie zur Verfügung steht. So entspricht ein NGS einem Multi-Gen-Panel mit einer Vielzahl von Markern (abhängig vom Anbieter) und es können Mutationen, Fusionen, Deletionen und Insertionen am Genom detektiert werden. Dies ist mit einer klassischen Sanger-Sequenzierung so nicht möglich. Beim NGS werden so alle krankheitsrelevanten Genveränderungen parallel untersucht. Es handelt sich um eine Hochdurchsatz-Sequenzierung.
Die folgende Tabelle gibt einen Überblick über therapeutisch relevante Biomarker. Aufgrund der schnellen Weiterentwicklungen in diesem Bereich erhebt sie jedoch keinen Anspruch auf Vollständigkeit.
| Marker | Erkrankung | Zielgerichtete Therapie |
| BCR / ABL CD 117 (c-kit) | CML GIST | Imatinib |
| CD-20 | B-NHL | Rituximab, Obinutuzumab, (Ofatumumab) |
| CD-30 | M. Hodgkin, T-NHL | Brentuximab-Vedotin |
| HER2/neu | Mamma- und Magenkarzinom* | Trastuzumab, Trastuzumab Emtansin (TDM1), Pertuzumab, Margetuximab Trastuzumab Deruxtecan Lapatinib, Neratinib, Tucatinib |
| PIK3CA | Mammakarzinom | Alpelisib |
| KRAS / NRAS KRAS | Kolonkarzinom NSCLC | Cetuximab / Panitumumab nicht wirksam! Orale TKI nicht wirksam! |
| KRAS G12C | NSCLC | Sotorasib |
| RET | NSCLC, Schilddrüsenkarzinom | Pralsetinib, Selpercatinib |
| EGFR Mutation | NSCLC | Afatinib, Dacomitinib, Erlotinib, Gefitinib, Osimertinib |
| EGFR T790M | NSCLC | Osimertinib |
| ALK | NSCLC | Alectinib, Brigatinib, Ceritinib, Crizotinib, Lorlatinib |
| ROS1 | NSCLC | Crizotinib, Entrectinib |
| NTRK | Tumoragnostischer Marker | Larotrectinib, Entrectinib |
| BRAF(BRAFV600) | Melanom Melanom + NSCLC Kolonkarzinom Haarzellenleukämie | Encorafenib + Binimetinib** Dabrafenib + Trametinib** Vemurafenib + Cobimetinib** Dabrafenib + Trametinib Encorafenib (+Cetuximab) |
| MSI | Rez. Endometriumkarzinom Kolonkarzinom Tumoragnostischer Marker in USA (FDA) | Dostarlimab, Pembrolizumab, Nivolumab+Ipilimumab |
| PD-1/PD-L1 | Nahezu alle soliden Tumore, Hodgkin-Lymphom | Atezolizumab, Avelumab, Cemiplimab, Durvalumab, Nivolumab, Pembrolizumab, Retifanlimab, Tislelizumab, Toripalimab |
| c-MET | NSCLC | Capmatinib, Tepotinib |
| BRCA 1 / 2 | Mammakarzinom, Pankreaskarzinom, Ovarialkarzinom***, Prostatakarzinom**** | Olaparib, Niraparib, Rucaparib, Talazoparib |
| HRD | Ovarialkarzinom, Mammakarzinom | Olaparib |
| FGFR2 oder-3 | mUrothelkarzinom Cholangiokarzinom | Erdafitinib Pemigatinib |
| * Beim Magenkarzinom nur Trastuzumab zugelassen ** Binimetinib, Cobimetinib und Trametinib sind MEK-Inhibitoren, keine BRAF-Inhibitoren *** Beim Ovarialkarzinom in der Zweitlinienerhaltungstherapie auch ohne BRCA-Mutation zugelassen **** nur Olaparib zugelassen | ||
2. Behandlungsverfahren
Als grundlegende Pfeiler der onkologischen Therapie gelten die Operation, die Strahlentherapie und die medikamentöse Tumortherapie. Diese Behandlungsmodalitäten finden entweder einzeln oder in Kombination Anwendung. Wird ein Patient im Rahmen des onkologischen Therapiekonzeptes mit mehreren dieser Behandlungsoptionen therapiert, so spricht man auch von multimodaler Therapie. Beispiele für multimodale Therapien sind die Behandlung lokal fortgeschrittener Bronchial-, Mamma- und Ösophaguskarzinome. Hier beginnt die onkologische Therapie zunächst mit einer zytostatischen Chemotherapie, gefolgt von einer kombinierten Chemostrahlentherapie und einer sekundären Tumorresektion.
2.1 Operative Therapie
Die operative Tumortherapie beschreibt die chirurgische Resektion von Tumoren mit einem ausreichenden Sicherheitsabstand. Die operative Therapie ist Grundlage vieler kurativer Therapiekonzepte. Bei den meisten Tumoren ist eine Kuration nur durch vollständige Entfernung des Tumors zu erzielen. Jedes entfernte Gewebe muss histopathologisch untersucht werden. Bei der Begutachtung durch den Pathologen ist insbesondere die Analyse der Schnittränder von entscheidender Bedeutung. Man unterscheidet allgemein drei verschiedene Resektionszustände:
| R0-Resektion | Makroskopisch und mikroskopisch komplette Entfernung des Tumors mit Sicherheitsabstand. |
| R1-Resektion | Makroskopisch komplette Tumorresektion, jedoch mikroskopisch verbliebene Residuen. Bei einer R1-Resektion beschreibt der Pathologe Tumorzellverbände, die bis an den Resektionsrand heranreichen. Auch ein Abstand der Tumorzellen von nur wenigen Millimetern zum Resektionsrand kann unter funktionellen und prognostischen Gesichtspunkten nicht mehr als R0-Resektion betrachtet werden, sodass auch hier von R1-Resektion gesprochen wird. |
| R2-Resektion | Makroskopisch verbliebene Tumorteile im Operationsgebiet. Bei diesem Resektionsstatus beendet der Chirurg die Operation in dem Wissen, dass noch Tumorreste verblieben sind. Bei manchen Tumoren wie Weichteilsarkomen werden R2-Resektionen zum Tumordebulking (Verminderung der bestehenden Tumorlast) angestrebt, um die Prognose einer späteren Strahlen- oder Chemotherapie zu verbessern. |
2.1.1 Weitere onkologisch-chirurgische Prinzipien/Maßnahmen
No-touch-Technik: Ein weiteres Grundprinzip der onkologischen Chirurgie ist es, den Tumor während der Resektion nicht zu berühren oder einzuschneiden. Damit soll eine intraoperative Streuung des Tumormaterials vermieden werden. Dies wird durch einen weiträumigen Sicherheitsabstand gewährleistet.
Lymphonodektomie: Die Entfernung der regionären und in einigen Fällen auch weiter entfernten Lymphknoten wird als Lymphonodektomie oder Lymphknotendissektion bezeichnet. Der Entfernung der regionären Lymphknoten kommt bei allen großen internistisch-onkologischen Tumorentitäten – Mamma-, Bronchial-, Magen- und Kolonkarzinom – eine besondere Bedeutung zu. Hier entscheidet insbesondere der Lymphknotenstatus (Zahl und Größe der befallenen Lymphknoten) über die weitere Therapie und die Prognose des Patienten.
Sicherheitsabstand: Ein weiteres onkologisch-chirurgisches Prinzip ist die Einhaltung weiträumiger Sicherheitsabstände, um insbesondere R1-Resektionen zu vermeiden. Dazu werden beispielsweise bei Resektionen von Darmtumoren Hemikolektomien durchgeführt, sodass mindestens ein Sicherheitsabstand von 5 cm oberhalb und unterhalb der Tumorgrenzen eingehalten werden kann. Bei der Resektion von Lebermetastasen gilt die Empfehlung, mindestens 1 cm im gesunden Gewebe zu resezieren.
Metastasenchirurgie: Die chirurgische Entfernung von Leber- oder Lungenmetastasen wird als Metastasektomie bezeichnet. Insbesondere bei Metastasen eines kolorektalen Karzinoms ist die Metastasenchirurgie etablierter Bestandteil der onkologischen Therapiekonzepte. Auch bei malignen Melanomen und Weichteilsarkomen kommt der Metastasenchirurgie prognostische Bedeutung zu.
Palliativoperationen: Manche chirurgischen Interventionen haben nicht zum Ziel, einen malignen Tumor vollständig zu entfernen. Diese Palliativoperationen dienen der Tumorverkleinerung (Debulking), um funktionell störende Einflüsse des Tumorkonglomerates zu minimieren. Typische Beispiele für Palliativeingriffe sind die Anlage von Gastroenterostomien bei einem total stenosierenden Magen- oder Dünndarmkarzinom oder auch einer sogenannten Witzel-Fistel zu Ernährungszwecken bei total stenosierendem Ösophaguskarzinom. Gleiches gilt für die Anlage eines Anus praeter bei diffuser peritonealer Karzinose und Ileus-Zuständen.
Organtransplantation: Prinzipiell gilt ein vorhandenes, aktives Malignom als Kontraindikation zur Organtransplantation. Dem liegt zugrunde, dass maligne Zellen unter der erforderlichen Immunsuppression deutlich schneller proliferieren als bei nicht unterdrückter Körperabwehr. In seltenen Fällen können jedoch anderweitig nicht operable Lebertumoren, z. B. hepatozelluläre Karzinome oder cholangiozelluläre Karzinome, die keine Fernmetastasen gebildet haben, eine Indikation zur Lebertransplantation darstellen.
Portsysteme: Auch die Anlage von venösen Portsystemen zur zytostatischen Chemotherapie oder zur supportiven Therapie ist Aufgabe des chirurgischen Onkologen. Unter einem Portsystem versteht man ein „Metallkästchen“, das unterhalb des Schlüsselbeines auf die Muskulatur genäht wird. Dieses Metallkästchen ist über einen Polyurethan-Katheter mit der oberen Hohlvene verbunden. Über dieses Portsystem können sowohl Blutentnahmen durchgeführt als auch zytostatische Behandlungen verabreicht werden (siehe auch Teil Durchführung Abschnitt Applikation). Auch supportive Maßnahmen wie Ernährungstherapien, Rehydratationen und intravenöse Schmerztherapien mittels Schmerzpumpen sind über Portsysteme möglich. Die zwei wesentlichen Komplikationen eines implantierten Portsystems sind die portassoziierte Venenthrombose und die Portinfektion mit Bakterien.
Entlastende Punktionen: Entlastende Punktionen von Aszites (Bauchwassersucht, Bauchfelltranssudat), Pleuraerguss (Brustfellerguss) oder Perikarderguss (Herzbeutelerguss) sind in der Regel keine chirurgischen, sondern internistische Aufgaben. Diese Eingriffe sind von kleinerem Umfang und stellen somit keine chirurgischen Maßnahmen im engeren Sinne dar. Trotzdem müssen auch diese Punktionen unter sterilen Kautelen durchgeführt werden.
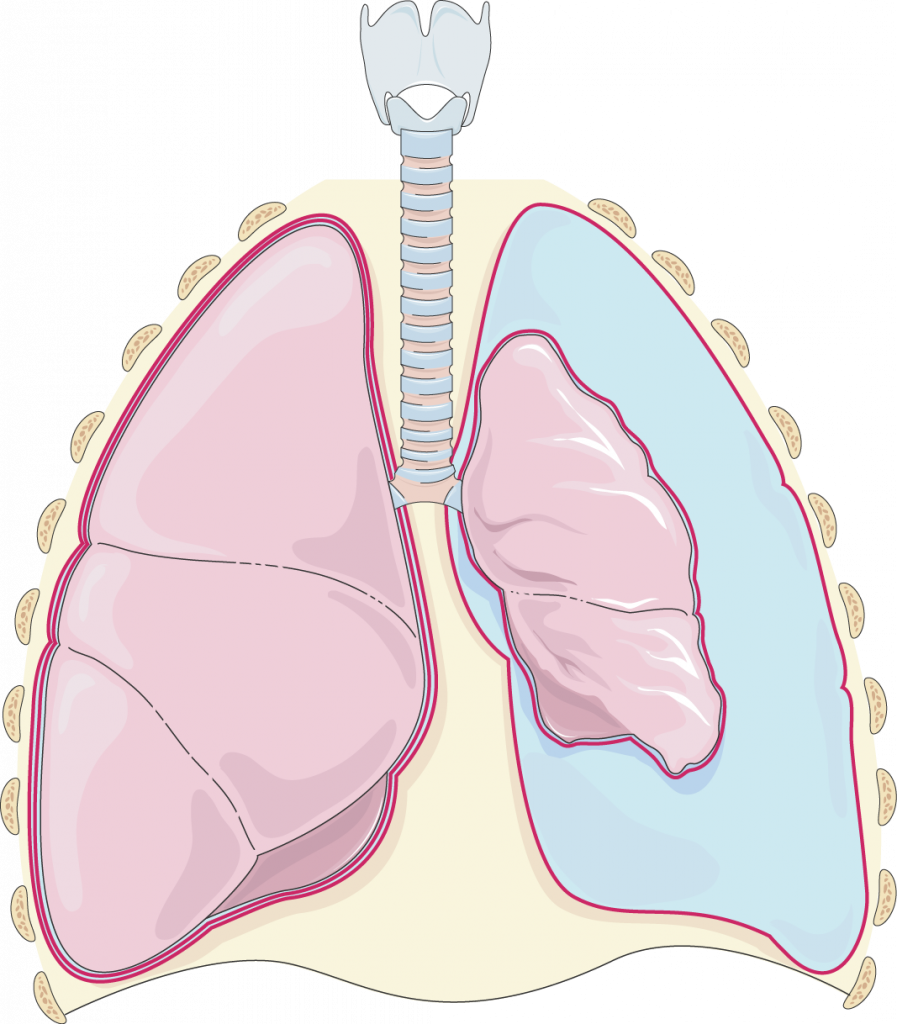
Die Anlage einer Aszites-, Pleura- oder Perikarddrainage erfolgt stets unter sonografischer Kontrolle zur Detektion des bestmöglichen Punktionsortes. Dabei muss durch die vorherige Ultraschalluntersuchung sichergestellt werden, dass keine Organe in der Nähe der Punktionsstelle verletzt werden können. Aszitesdrainagen werden nach Ablassen einer bestimmten Aszitesmenge am Punktionstag entfernt. Ein permanent einliegender Katheter in der Bauchhöhle ist nicht erforderlich.
Die Anlage eines Katheters über mehrere Tage kann jedoch bei Perikard- oder Pleurapunktionen erforderlich werden. Dies ist insbesondere der Fall, wenn eine komplette Drainage der serösen Höhle erreicht werden muss, was durch Sogunterstützung erfolgen kann. Die komplette Drainage ist immer vor einer Pleurodese oder Perikardiodese erforderlich (siehe auch Abschnitt Weitere Chemotherapieformen).
Für Perikard- und Pleurapunktionen stehen zusammengestellte sterile Sets zur Verfügung. Unter Notfallbedingungen erfolgt die Punktion mit einer großlumigen Venenverweilkanüle. Im Gegensatz zu den fertigen Sets für Pleura- und Perikardpunktionen müssen die Materialien für eine Aszitespunktion, in seltenen Fällen auch für eine Pleurapunktion, einzeln zusammengestellt werden.
Materialien für eine Aszitespunktion: Desinfektionsmittel, sterile Tupfer, sterile Pflaster, sterile Venenverweilkanüle 1,4 mm Durchmesser, Ablaufbeutel mit sterilen Konnexionsüberleitungen, Drei-Wege-Hahn, fakultativ Lokalanästhetika wie Scandicain zur örtlichen Betäubung, Pflaster zum Fixieren der Ablaufleitungen, Unterlagen zur Schonung des Bettzeugs (Pleurapunktionen und Aszitesdrainagen erfolgen in der Regel unter Ultraschall im Krankenbett), sterile Handschuhe, Seitenlage, Sandsack, um ein Nachlaufen des Punktats zu verhindern. Sterile Kautelen sind wichtig, um eine sekundäre Infektion der serösen Höhle zu vermeiden.
2.2 Strahlentherapie
2.2.1 Physikalisches Prinzip
Unter einer Strahlentherapie versteht man die Behandlung des malignen Tumors mittels radioaktiver Strahlen. Aus physikalischer Sicht handelt es sich bei den radioaktiven Strahlen um beschleunigte Elektronen. Sie werden als Korpuskularstrahlen bezeichnet. Daneben können Strahlentherapien auch mit Photonenstrahlen (= elektromagnetische Wellen) erfolgen.
Die Erzeugung der radioaktiven Strahlen erfolgt in Linearbeschleunigern oder Kreisbeschleunigern. Dabei liegt bei der Entstehung der Strahlung das gleiche physikalische Prinzip zugrunde wie bei der Entstehung von Röntgenstrahlen. Es wird durch Erhitzen eines Glühwendels eine Elektronenwolke gebildet, die dann durch Anlegen einer Spannung vom negativen Pol (sogenannte Kathode) zum positiven Pol (sogenannte Anode) beschleunigt wird. Es entsteht ein hochenergetischer Elektronenstrahl. Dieser prallt auf die aus Wolfram bestehende Anode, sodass hier potenzielle Energie in Form von Röntgenstrahlung freigesetzt wird. Diese Strahlung wird gebündelt und somit auf ein definiertes Feld, das sogenannte Strahlenfeld, eingestrahlt.
2.2.2 Biologische Wirkungen
Die radioaktiven Strahlen üben durch Ionisation und Anregung von Atomen eine Energieübertragung auf die Zielzellen (= Tumorzellen) aus. Dabei wird die Eindringtiefe in das Gewebe durch die Strahlenart und die Energiedosis bestimmt. Im Gewebe selbst werden sowohl Radikale mit positiven und negativen Ladungen als auch freie Elektronen gebildet.
Dadurch werden chemische Reaktionen initiiert, die die Desoxyribonukleinsäure (DNS), die Proteine der Zelle und auch die Zellmembran schädigen. Durch die Einwirkung der radioaktiven Strahlen auf die DNS kommt es zu Strangbrüchen, sodass die Replikation der DNS und auch die Transkription zur RNS unterbrochen werden. An der DNS bewirkt die radioaktive Strahlung somit eine punktförmige Schädigung (sogenannte Treffertheorie).
Dabei ist der Schaden an der Zelle umso größer, je mehr „Treffer“ durch die radioaktive Strahlung gesetzt worden sind. Das gleichzeitige Eintreten mehrerer Treffer wird als Konzentrationseffekt bezeichnet. Dies ist ein biologisches Ereignis mit ausgeprägter zellschädigender Wirkung, sodass sogar Chromosomenbrüche induziert werden können.
Neben dieser direkten Wirkung an der DNS ist die radioaktive Strahlung auch über indirekte Wirkungen aktiv. Diese indirekten Wirkungen beruhen auf der Aktivierung von Atomen in der Umgebung einer Zelle, sodass die Zellschädigung nicht direkt durch die Strahlung, sondern über gebildete reaktive Verbindungen „in der Nachbarschaft“ ausgelöst wird.
Unter strahlenbiologischen Aspekten gilt grundsätzlich, dass schnell proliferierende Zellen durch eine Bestrahlung stärker geschädigt werden als ruhende Zellen.
Zellen in der Mitosephase des Zellzyklus sind besonders strahlenempfindlich und eine intrazelluläre Reparatur solcher Schäden ist in dieser Zellzyklusphase kaum möglich. In der sogenannten G1-Phase sind die Zellen relativ strahlensensibel. Im weiteren Verlauf der Synthesephase steigt die Strahlenresistenz der Zellen an, weil in diesen Phasen die Reparaturvorgänge besonders ausgeprägt sind.
Nach Abschluss der DNS-Synthese und der DNS-Replikation sind die Zellen erneut so strahlenempfindlich wie in der Mitosephase. Die biologischen Wirkungen einer radioaktiven Bestrahlung sind dabei von der Strahlendosis abhängig. So wird bei einer niedrigen Strahlendosis zunächst die DNS-Synthese von Zellen herabgesetzt. Mit zunehmender Strahlendosis werden weitere irreversible Schäden in der Zelle gesetzt, die zu untypischen Teilungsprozessen und zur Zytolyse führen.
2.2.3 Strahlensensibilität der Tumoren
Die biologische Strahlenempfindlichkeit von Malignomen ist sehr unterschiedlich. Als hochgradig strahlensensibel gelten Leukämien, hochmaligne Lymphome, das Chorionkarzinom, das Seminom, die Embryonaltumoren und das kleinzellige Bronchialkarzinom. Als mäßiggradig strahlensensibel werden Plattenepithelkarzinome, das Osteosarkom, Glioblastome, die Adenokarzinome und die Teratokarzinome angesehen. Demgegenüber sind ausgereifte Sarkome und alle benignen Tumoren strahlenresistent. Auch das maligne Melanom gilt trotz schnellen Wachstums als strahlenresistenter Tumor.
Die Strahlenwirkung auf den malignen Tumor hängt von der verabreichten Strahlendosis ab; die applizierbare Strahlendosis wiederum ist insbesondere von den an den Tumor grenzenden Nachbargeweben „abhängig“. Bei jeder Strahlentherapie wird auch gesundes Gewebe im Sinne eines Sicherheitsabstandes durch die Bestrahlung geschädigt. Dabei kann grundsätzlich keine höhere Strahlendosis verabreicht werden, als im umgebenden Gewebe tolerabel ist. Weiterhin ist strahlenbiologisch bedeutsam, dass nicht die gesamte erforderliche tumorizide Dosis in einer Fraktion (= als Einmalgabe) verabreicht werden kann. Die Gesamtdosis muss zeitlich verteilt werden, was als Fraktionierung bezeichnet wird.
2.2.4 Bestrahlungsformen
Als grundlegende Bestrahlungsform gilt bei malignen Tumoren die perkutane Bestrahlung, das heißt die Bestrahlung des Tumors von extern durch die Haut. Dabei muss vor jeder Radiatio eine Strahlenplanung erfolgen, wobei das bestrahlte Feld unter computertomografischer Kontrolle berechnet und mit wasserfester Farbe auf der Körperhaut markiert wird. Dann erst erfolgt die Abgabe der radioaktiven Strahlung von einer feststehenden Röntgenröhre aus. Dieses Vorgehen wird als Stehfeldbestrahlung bezeichnet.
Bei oberflächlich gelegenen Tumoren ist es meist ausreichend, den Tumor aus einer Richtung bzw. über ein Feld zu bestrahlen, als Einfeldbestrahlung bezeichnet.
Bei tiefer gelegenen Tumoren erfolgt die Bestrahlung über verschiedene Felder, um Toxizität im gesunden Gewebe einzusparen. Man spricht hier von Mehrfeldbestrahlung. Handelt es sich dabei um zwei Strahlenfelder, die einander genau gegenüber liegen (opponierende Stehfelder), wird auch von einer Gegenfeldbestrahlung gesprochen. Erfolgt die Bestrahlung aus mehr als zwei Feldern, wird der Terminus Kreuzfeuerbestrahlung verwendet. Wird die Strahlung aus einer beweglichen Röntgenröhre abgegeben, so spricht man von Rotationsbestrahlung oder Pendelbestrahlung. Im Gegensatz zu den vorher genannten Bestrahlungstechniken besteht die Pendelbestrahlung aus einer maximalen Vielzahl an Einzelstehfeldern.
Eine Sonderform der Bestrahlung ist die sogenannte intrakavitäre Therapie (synonym Brachytherapie). Darunter versteht man die Bestrahlung eines malignen Tumors, indem das strahlende Nuklid in eine Körperhöhle eingeführt wird. Beispiele für intrakavitäre Therapien sind die After-loading-Bestrahlung bei Zervixkarzinomen oder die Bestrahlung von Ösophaguskarzinomen durch eine direkt im Ösophagus einliegende Strahlungsquelle. Bei einer intrakavitären Therapie können Strahler mit einer höheren Dosisleistung verwendet werden, als dies bei einer perkutanen Bestrahlung der Fall ist.
2.2.5 Nuklearmedizinische Verfahren
Neben den reinen Bestrahlungstherapien können in nuklearmedizinischen Abteilungen Behandlungen mit sogenannten offenen Radionukliden erfolgen. Bekanntestes Beispiel dafür ist die Radiojodtherapie bei Schilddrüsenkarzinomen. Prinzip der offenen Radionuklidtherapie ist die intravenöse Applikation eines radioaktiven Pharmakons, welches sich über den Blutweg selektiv im zu bestrahlenden Gewebe anreichert. Dies ist bei den Schilddrüsenkarzinomen durch ihre Fähigkeit zur Jodspeicherung in besonderer Weise ausgeprägt. Auch können Knochenmetastasen mit 89Strontium behandelt werden, z. B. beim Prostatakarzinom.
Unter einer Seedimplantation (Seedtherapie, interstitielle Bestrahlung) versteht man allgemein das Einbringen eines oder mehrerer radioaktiv markierter Stäbchen in ein Tumorareal, sodass eine „innere“ Bestrahlung auf einem möglichst kleinen Raum und somit mit möglichst wenig Nebenwirkungen erfolgt. Da die verwendeten Strahler nur eine kurze Reichweite im Gewebe haben, müssen mehrere Seeds in genau definierten Abständen in das tumortragende Areal eingebracht werden, was in der Regel in Narkose geschieht. Klinische Versuche beziehen sich insbesondere auf das Prostatakarzinom, bei dem lange progressionsfreie Intervalle an selektionierten Patientengruppen beschrieben sind.
Eine weitere nuklearmedizinische Behandlungsmöglichkeit ist die „Selektive Interne Radiotherapie“ (SIRT oder Radioembolisation), bei der radioaktive Kügelchen (Beta-Strahler), sog. Mikrosphären mit einem Durchmesser von 35 µm, in das Tumorgewebe mithilfe eines Katheters eingebracht werden. Diese Mikrosphären bleiben in kleinen Tumorgefäßen „stecken“ und bewirken eine interne Bestrahlung. SIRT kann bei primären Lebertumoren oder Lebermetastasen angewendet werden, wenn konventionelle Standardtherapien nicht möglich sind.
2.2.6 Strahlensensibilisierende Möglichkeiten (Radiosensitizer)
Durch Applikation bestimmter Medikamente ist es in Einzelfällen möglich, die Wirksamkeit der radioaktiven Bestrahlung zu verstärken. So kann der Effekt der Radiotherapie modellhaft erhöht werden durch:
- Re-Oxygenierung der Tumorzellen (Hypoxie ist ein Resistenzmechanismus, der Tumorzellen vor Strahlenschäden schützt)
- Inhibition der Reparaturmechanismen bestrahlter Tumorzellen
- Aktivierung der Tumorzellen aus der G0-Phase (Ruhephase) des Zellzyklus
- Verhinderung der Regeneration von Tumorzellen
Diese Effekte macht man sich bei einer kombinierten Strahlenchemotherapie zunutze. Medikamente wie Cisplatin und Doxorubicin sind beispielsweise strahlensensibilisierende Substanzen. Als Mechanismus gilt dabei die Arretierung der Zellen in einer strahlensensiblen Phase des Zellzyklus. Zudem verhindern Cisplatin oder Doxorubicin die Reparatur subletaler Strahlenschäden an den bestrahlten Zellen.
2.2.7 Nebenwirkungen der Strahlentherapie
Wie bereits erwähnt, wird durch jede Bestrahlung neben dem neoplastischen Gewebe auch gesundes Körpergewebe geschädigt. Das sogenannte Strahlenerythem ist ein typisches Beispiel einer Hautschädigung, die Haut wird bei zunehmender Bestrahlungsdauer rot. In der Zeit vor der exakten Dosimetrie wurde die Erythembildung herangezogen, um zu bestimmen, zu welchem Zeitpunkt die entsprechend wirksame Dosis appliziert war (sogenannte Erythemdosis).
Prinzipiell sind Strahlenschäden an jedem bestrahlten Organ möglich. Dabei gilt das blutbildende Knochenmark als sehr empfindliches Organsystem. Auch die während der Bestrahlung durch das Bestrahlungsfeld fließenden Blutbestandteile, insbesondere Leukozyten, werden durch die Bestrahlung geschädigt. Typische Nebenwirkungen bei einer Bestrahlung im Bereich des Thorax sind die Lungenfibrose im bestrahlten Feld bzw. die radiogene Perikarditis, falls Anteile des Perikards im Strahlenfeld liegen. Im Bereich des Abdomens kann es zu einem Untergang der Leberzellen bei der Bestrahlung lebernah gelegener Tumoren kommen. Befindet sich eine Niere im Strahlenfeld, ist zu erwarten, dass die Filtrationsleistung der Niere abnimmt. Im Bereich des Intestinaltraktes kommt es zu Schädigungen der Dünndarmepithelien mit der klinischen Konsequenz einer radiogenen Diarrhö. Bei Bestrahlungen im Kopf-Hals-Bereich kann es zur Kataraktbildung kommen. Bei höheren Dosen können neurologische Schädigungen auftreten. Beim Überschreiten der zulässigen Toleranzgrenze des Rückenmarks kann das Maximalbild eines radiogenen Querschnitts resultieren.
Neben diesen akuten Nebenwirkungen sind auch Spätschäden durch die Bestrahlung, insbesondere die Induktion von Zweitneoplasien zu nennen. So können 5–20 Jahre nach Abschluss einer Strahlentherapie im bestrahlten Areal Weichteilsarkome auftreten. Durch die Bestrahlung des blutbildenden Knochenmarks kann es zu sekundären Leukämien kommen.
2.3 Medikamentöse Tumortherapie
Die konservative medikamentöse antineoplastische Therapie umfasst die Behandlung von Tumoren mit klassischen Zytostatika, Hormonen, Antikörpern oder immunmodulatorisch wirksamen Substanzen.

Dabei wird der Begriff „medikamentöse Tumortherapie“ vielfach mit dem Begriff „Chemotherapie“ gleichgesetzt, der allerdings nicht auf die antineoplastische Behandlung allein beschränkt ist. Auch die Behandlung bakterieller Infektionen mit Antibiotika wird als Chemotherapie bezeichnet. Darüber hinaus wird der Begriff „Chemotherapie“ vielfach auch gleichgesetzt mit dem Begriff „Zytostatikatherapie“. Im strengeren Sinne ist jedoch der Begriff „zytostatische Chemotherapie“ nicht korrekt, weil die verabreichten Medikamente einen Tumorzelltod induzieren, also zytotoxisch wirksam sind. Der Begriff „zytostatisch“ bedeutet demgegenüber, dass die verabreichten Medikamente das Wachstum von Tumorzellen inhibieren, jedoch keinen Zelltod induzieren können.
Eine zytostatische Chemotherapie kann unter verschiedenen Gesichtspunkten erfolgen: So kann die zytostatische Chemotherapie bei bestimmten Neoplasien in kurativer Absicht erfolgen, während sie bei weit fortgeschrittenen Malignomen lediglich in palliativer Intention eingesetzt wird. Eine zytostatische Chemotherapie kann nach einer Tumorresektion erfolgen (adjuvant oder additiv) oder einer kurativen Behandlung vorgeschaltet sein (neoadjuvant). Dabei werden der Ablauf und die Intensität der zytostatischen Chemotherapie von der zugrunde liegenden Intention bestimmt.
Chemotherapien können grundsätzlich als Monochemotherapie oder als Polychemotherapie angewendet werden.
2.3.1 Prinzipien der zytostatischen Chemotherapie
Physiologisches Prinzip einer zytostatischen Chemotherapie ist es, rasch proliferierende Zellen selektiv anzugreifen. Dem liegt zugrunde, dass Zytostatika in schnell proliferierende Zellen vermehrt inkorporiert werden. Wichtig ist dabei jedoch zu wissen, dass es keine 100%-ige Selektivität gibt und durch jede zytostatische Chemotherapie immer gesunde Zellen geschädigt werden. Dabei ist die Effizienz der zytostatischen Chemotherapie umso größer, je höher das proliferative Potenzial des zu behandelnden Tumors ist. So werden undifferenzierte (G3) Tumoren von einer zytostatischen Chemotherapie deutlich stärker getroffen als hochdifferenzierte (G1) Tumoren. Folgende allgemeine Differenzierungsgrade maligner Tumoren (Grading)[1] werden unterschieden:
| G1 | hochdifferenzierter Tumor (niedrige Proliferationsrate, geringe Chemosensibilität) |
| G2 | mäßiggradig differenzierter Tumor |
| G3 | niedrigdifferenzierter Tumor (hohe Proliferationsrate, hohe Chemosensibilität) |
| G4 | anaplastischer (= undifferenzierter) Tumor |
Jede zytostatische Chemotherapie setzt einerseits eine Toxizitätskontrolle und andererseits eine Erfolgskontrolle voraus. Dies bedeutet, dass zu Beginn jeder zytostatischen Chemotherapie messbare Parameter bezüglich der Tumorausbreitung erhoben werden müssen. Dazu können Ultraschalluntersuchungen, Röntgenuntersuchungen oder Computertomografien von der betroffenen Region durchgeführt werden. Diese haben zum Ziel, den Tumor mindestens in einer Ebene darzustellen. Auch Tumormarker können zur Erfolgskontrolle einer zytostatischen Chemotherapie eingesetzt werden, vorausgesetzt, dass zu Therapiebeginn erhöhte Tumormarkerwerte nachgewiesen werden können. Einzige Ausnahme, bei der ohne jede messbare Läsion und ohne erhöhte Tumormarker therapiert wird, ist die adjuvante zytostatische Chemotherapie.
Weiter sind Patienten im Rahmen der Toxizitätskontrolle regelmäßig auf Nebenwirkungen zu befragen. Die relevanten Organfunktionen sind vor Therapiebeginn im Sinne eines Basisstatus zu untersuchen. Beispiele für die Toxizitätskontrolle sind regelmäßige echokardiografische Untersuchungen unter Therapie mit kardiotoxischen Anthrazyklinen oder regelmäßige Lungenfunktionsprüfungen unter Therapie mit Bleomycin, um Lungenfibrosen und Pneumonitiden frühzeitig zu erkennen.
Zu Beginn einer onkologischen Therapie müssen alle vorhandenen Tumormanifestationen bildgebend dokumentiert werden. Hierauf basiert im Verlauf die Evaluation des Tumoransprechens.
2.3.1.1 Remissionsdefinitionen (RECIST, Version 1.1)
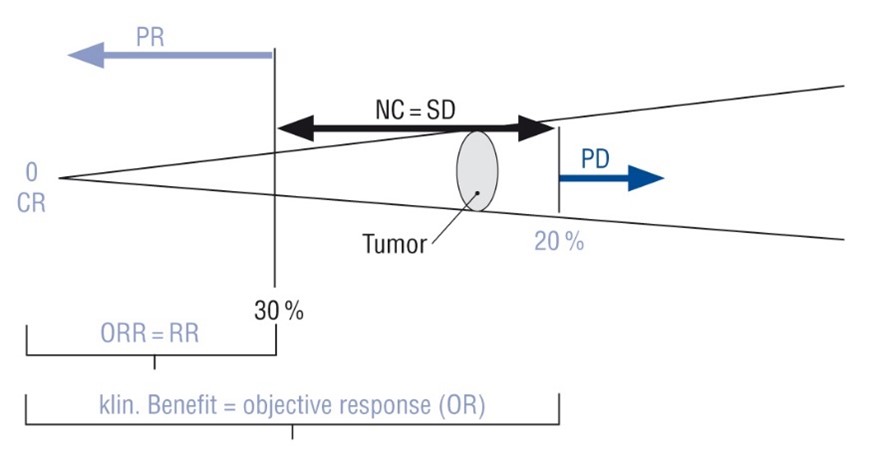
Die Tumorgröße ist definiert als „Summe der Durchmesser der Zielläsionen (target lesions)“.
| Komplette Remission (CR) | Sind nach einer antineoplastischen Therapie alle tumorbedingten Veränderungen über einen Zeitraum von mindestens 4 Wochen nicht mehr nachweisbar, so spricht man von kompletter Remission. |
| Partielle Remission (PR) | Eine Rückbildung der Tumorgröße im Vergleich zum Ausgangsbefund um 30% oder mehr wird als partielle Remission bezeichnet. Dabei kann die Messung der Tumormanifestation zweidimensional erfolgen (WHO-Kriterien) oder eindimensional (RECIST-Kriterien, RECIST = Response Evaluation Criteria In Solid Tumors). |
| No Change (NC, SD) | Eine gleichbleibende Tumormanifestation wird als „no change“ oder „stable disease“ bezeichnet. Dabei hat die Tumormanifestation, verglichen mit dem Ausgangsbefund, um weniger als 20% zu- bzw. weniger als 30% abgenommen. |
| Progression (PD) | Eine Größenzunahme des Tumors um mehr als 20% seiner ursprünglichen Größe oder das Auftreten neuer Tumormanifestationen wird als Progression bezeichnet. |
| Diskordantes Ansprechen | Bildet sich bei Vorhandensein mehrerer Tumormanifestationen oder mehrerer Metastasen eine Manifestation zurück, während gleichzeitig andere Manifestationen oder Metastasen an Größe zunehmen bzw. neue Metastasen auftreten, so liegt ein diskordantes Ansprechen vor. Jedes diskordante Ansprechen ist jedoch zuungunsten des Patienten als Progression zu werten. |
2.3.2 Monochemotherapie
Eine zytostatische Chemotherapie mit einem einzigen Zytostatikum wird als Monotherapie bezeichnet. Die Therapie basiert somit auf einem bestimmten Wirkstoff, wie beispielsweise Trifluridin. Eine Monotherapie findet häufig in palliativen Therapiesituationen Anwendung. Dabei wird zugrunde gelegt, dass eine Monotherapie grundsätzlich weniger toxisch ist als eine zytostatische Kombinationstherapie (siehe Abschnitt Polychemotherapie). Dementsprechend ist auch die Remissionsrate unter einer Monochemotherapie meist niedriger als unter einer Kombinationstherapie. Monochemotherapien werden beispielsweise bei Plasmozytomen, bei chronischen myeloischen Leukämien oder auch beim kleinzelligen Lungenkarzinom eingesetzt. Darüber hinaus gilt weiterhin, dass Dritt- oder Viertlinientherapien, um unverhältnismäßige Toxizitäten zu verhindern, monotherapeutisch erfolgen sollen.
2.3.3 Polychemotherapie
Die Kombination mehrerer zytostatisch aktiver Substanzen in der antineoplastischen Behandlung wird als Polychemotherapie bezeichnet. Dabei wird durch den kombinierten Einsatz eine höhere Remissionsrate als im Vergleich zur Monotherapie erreicht. Eine höhere Toxizität wird in Kauf genommen. Polychemotherapien finden meist in kurativen Therapiekonzepten Anwendung. Beim Einsatz einer Polychemotherapie ist bei der Auswahl der Medikamente darauf zu achten, dass einerseits unterschiedliche Wirkmechanismen vorliegen, um den Tumor über verschiedene Stoffwechselwege anzugreifen. Darüber hinaus ist weiter darauf zu achten, dass sich die Nebenwirkungen der verabreichten Zytostatika nicht überlappen. Somit ist es Sinn einer Kombinationstherapie, eine höhere Remissionsrate bei möglichst nicht überschneidender Toxizität zu erzielen. Dadurch werden primär resistente Tumorzellen mit einer höheren Wahrscheinlichkeit erreicht als bei einer Monochemotherapie. Auch die Entwicklung sekundär resistenter Zellklone erfolgt bei Kombinationstherapien langsamer als bei Monochemotherapien. Vom onkologischen Prinzip her dürfen in Kombinationstherapien jedoch nur Medikamente verwendet werden, die auch monotherapeutisch bei der zu behandelnden Grunderkrankung aktiv sind.
2.3.4 Zeitliche Abfolge der Polychemotherapie
Jürgen Barth
Die ersten effektiven Substanzen gegen Krebs kamen Mitte bis Ende der 1940er Jahre zum Einsatz, die anfänglichen therapeutischen Erfolge der Monotherapie waren jedoch enttäuschend (partielle Remissionen von kurzer Dauer). Die Polychemotherapie wurde zirka 10 Jahre später zuerst gegen die kindliche akute lymphatische Leukämie ALL und gegen Lymphome etabliert und stellte einen Wendepunkt der Krebsbehandlung dar.
Im Rahmen der Polychemotherapie versucht man, synergistische Effekte der antitumoral wirkenden Einzelsubstanzen zu erzielen, wobei die Organtoxizitäten nicht überlappend sein sollten. Anfänglich hoffte man, die Tumorzellen in ihrem Zellzyklus „synchronisieren“ zu können, indem man zuerst einen Mitosehemmstoff wie ein Vinca-Alkaloid appliziert, wodurch die Zellen vor ihrer Teilungs-/Synthesephase arretiert werden. Anschließend erfolgt die weitere Chemotherapie, beispielsweise mit phasenunspezifischen Alkylanzien oder phasenspezifischen Antimetaboliten. Diese „Badewannenpharmakologie“ war jedoch nicht erfolgreich, da die Arzneistoffe nicht gleichmäßig und nicht in alle gewünschten Gewebe an- und abfluten. Die Gründe für die Ungültigkeit dieser Theorie sind somit pharmakokinetisch, biochemisch und, wie man heute weiß, substanzabhängig auch pharmakogenetisch bedingt. Es gibt jedoch agonistische und antagonistische „Interaktionen“ in Abhängigkeit der Applikationssequenz der Einzelsubstanzen.
Eine Verstärkung der antitumoralen Aktivität sieht man z. B., wenn Methotrexat (MTX) vor Fluorouracil (5-FU) gegeben wird. Im umgekehrten Fall (5-FU vor MTX) wird die Antifolatwirkung durch 5-FU abgeschwächt. Die MTX-Wirkung wird natürlich durch eine Gabe von Folaten vor MTX außer Kraft gesetzt, während die Applikation vor oder während einer 5-FU-Gabe die Toxizität erhöht.
Beispiele für bekannte Synergismen und Antagonismen einer Kombinationschemotherapie:
Verstärkung der Antitumoraktivität
- MTX vorab erhöht die 5-FU-Aktivierung
- MTX vorab erhöht die Ara-C-Aktivierung
- Folinate verstärken die Hemmung der Thymidylatsynthetase, wenn sie vor oder mit 5-FU gegeben werden
- Inhibitoren der Pyrimidin-de-novo-Synthese erhöhen den Einbau von 5-FU in RNS und die Bildung aktiver Nukleotide
- Synergismus der Sequenz Paclitaxel vor Cisplatin, sonst kommt es zu abschwächenden Effekten
- Jedoch Paclitaxel nach Anthrazyklin (Doxorubicin), weil in umgekehrter Reihenfolge die Myelotoxizität des Anthrzyklins dramatisch zunimmt (mechanistisch nicht verstanden; s.u.).
Abschwächung der Antitumoraktivität
- Vorbehandlung mit 5-FU vermindert die MTX-Aktivität
- Vorbehandlung mit Asparaginase blockiert die MTX-Effekte
- Cisplatin vor Paclitaxel reduziert die Taxanwirkung
- Antagonismus, wenn Paclitaxel nach Cisplatin gegeben wird (siehe oben)
- unspezifische Zerstörung von Zytochrom P 450, beispielsweise durch Cisplatin, könnte – theoretisch – die Aktivierung der Oxazaphosphorine (Cyclo-, Ifosfamid) unterbinden (Unterdosierung)
Besonderheit: Taxane in Kombination mit Anthrazyklinen
Wird ein Taxan mit einem Anthrazyklin kombiniert, dann sollte das Anthrazyklin vor dem Taxan gegeben werden. In umgekehrter Reihenfolge resultieren erhöhte Plasmaspiegel des Anthrazyklins und dessen Metabolite. Ohne dass der Mechanismus hierfür bekannt ist, resultieren aus der verminderten Ausscheidung verstärkte Nebenwirkungen wie Stomatitis und/oder (febrile) Neutropenien. Die Fachinformation von Paclitaxel empfiehlt sogar eine Gabe von Paclitaxel 24 h nach Doxorubicin.
Es soll nicht unerwähnt bleiben, dass die Reihenfolge der Verabreichung von Zytostatika auch völlig belanglos – also variabel – sein kann. Wissenschaftlich gesicherte Daten dazu gibt es, von den oben erwähnten abgesehen, kaum. Man sollte sich jedoch vergegenwärtigen, dass die in Publikationen angegebenen Ansprechraten mit einer bestimmten Medikamentenreihenfolge erzielt wurden. Ein eigenmächtiges Abändern der Sequenz, z. B. aus Gründen der Praktikabilität, kann nicht empfohlen werden, solange nachteilige Interaktionen nicht zweifelsfrei ausgeschlossen werden können.
2.3.5 Weitere Chemotherapieformen
Norbert Schleucher
Salvage-Chemotherapie: Eine Fortführung der zytostatischen Chemotherapie nach Versagen einer primären Standardtherapie wird als Salvage-Chemotherapie oder auch als Zweitlinientherapie bezeichnet. Dabei kommen im Rahmen von Therapien nicht nur etablierte Zytostatika zum Einsatz, sondern unter Umständen auch Medikamente, die klinisch noch nicht erprobt sind. Bei Salvage-Chemotherapien sind die zu erwartenden Remissionsraten in der Regel niedriger als bei den verwendeten Erstlinientherapien. Salvage-Therapien bedürfen – wie alle Therapieformen – der Aufklärung/Einwilligung der Patienten.
Dauertherapie: Eine zytostatische Chemotherapie, bei der die Medikamente kontinuierlich verabreicht werden, wird als Dauertherapie bezeichnet. Dabei kommen meistens oral verfügbare Zytostatika zum Einsatz. Beispiele für den Einsatz von Dauertherapien sind niedrigmaligne Non-Hodgkin-Lymphome (Dauertherapie mit der Substanz Chlor-ambucil), Weichteilsarkome (Dauertherapie mit Trofosfamid) oder myeloproliferative Syndrome. Prinzipiell ist die Dauertherapie eine selten angewendete Therapieform.
Stoßtherapie: Das Verabreichen der zytostatischen Chemotherapie in Intervallen wird sowohl bei einer Mono- als auch bei einer zytostatischen Polychemotherapie als Stoßtherapie bezeichnet. Bei einer Stoßtherapie werden die Zytostatika am Anfang des Therapiezyklus über einen oder mehrere Tage verabreicht. Danach erfolgt ein therapiefreies Intervall. Die meisten Chemotherapieregime im Rahmen von Stoßtherapien werden in drei- bis vierwöchigen Intervallen wiederholt. In der therapiefreien Zeit sind Blutbildkontrollen erforderlich, da Blutbildveränderungen im Rahmen von Stoßtherapien meistens erst 10–12 Tage nach Therapiestart auftreten (Nadir). Dabei ist der nächste Therapiezyklus erst nach Restitution der Blutwerte möglich.
Lokoregionäre Chemotherapie: Die isolierte zytostatische Behandlung eines Organs oder einer Extremität wird als lokoregionäre Chemotherapie bezeichnet. Beispiele für lokoregionäre Chemotherapien sind die Chemoperfusion der Leber bei einer inoperablen isolierten Lebermetastasierung. Auch die Extremitätenperfusion bei Weichteilsarkomen oder malignen Melanomen stellt eine lokoregionäre Chemotherapie dar. Dabei wird die zytostatische Chemotherapie direkt in die tumorversorgende Arterie appliziert. Insbesondere bei der Leberperfusion ist dabei die Applikation der zytostatischen Chemotherapie über ein intraarteriell implantiertes Portsystem möglich. Vorteil einer lokoregionären Chemotherapie ist es, in kurzer Zeit eine hohe Zytostatikakonzentration direkt in das befallene Gewebe zu applizieren. Die so verabreichte Substanz hat keine metabolischen Veränderungen durch die Leberpassage erfahren. Eine lokoregionär angewendete Chemotherapie wird erst nach der Tumorpassage hepatisch metabolisiert und eliminiert (siehe auch Teil Durchführung Abschnitt Lokoregionale Applikation).
Weitere lokoregionäre Therapieverfahren: Neben der oben genannten Chemoperfusion sind auch die Chemoembolisation und die radiofrequenzinduzierte Thermoablation (RITA) zu den lokoregionären Therapieverfahren zu zählen.
Als Chemoembolisation bezeichnet man das Einbringen von „Fremdmaterial“ in ein großes Gefäß, das die arterielle Versorgung eines Tumors oder einer Metastase sicherstellt. Methodisch wird dabei ein arterieller Katheter über die Bein- und Beckenarterien durch die Aorta bis in das tumorversorgende Gefäß vorgeschoben (sog. transarterielle Chemoembolisation, TACE). Anschließend erfolgt die Applikation einer chemischen Verbindung, z. B. Lipiodol in Kombination mit einem Zytostatikum wie Doxorubicin oder Cis-platin, zur „Verstopfung“ des zuführenden Gefäßes. Dadurch wird die Sauerstoff- und Nährstoffzufuhr des Tumors oder der Metastase gestoppt. Da die Chemoembolisation einen sofortigen Verschluss des versorgenden Gefäßes bedeutet, kommt es nicht durch Kollateralisierung zur Sicherstellung der Nährstoffversorgung wie bei einem langsamen Verschluss. Im embolisierten Bereich wird dann das gekoppelte Zytostatikum freigesetzt. Folge ist eine Nekrotisierung der Tumorzellen.
Eine Verbesserung bzw. Weiterentwicklung der TACE gelang durch die Anwendung medikamentenbeladener Mikropartikel, sog. „drug-coated beads (dc beads)“, auf deren Oberfläche das Zytostatikum Doxorubicin aufgrund elektrostatischer Kräfte haftet und damit noch selektiver an das Tumorgewebe abgegeben wird als bei einer „einfachen“ TACE. Somit gilt in Westeuropa die TACE mit dc beads als Standardverfahren (siehe auch Teil Durchführung Abschnitt Lokoregionale Applikation).
Unter einer radiofrequenzinduzierten Thermoablation (RITA) versteht man ein Verfahren zur Koagulation von Metastasen oder Tumoren. Dabei wird unter computertomografischer Kontrolle ein Ablationskatheter in das Zentrum der Metastase vorgeführt und anschließend durch Applikation von Radiofrequenzwellen Hitze erzeugt. Es resultiert eine thermische Koagulation der Metastase. Eine Rita ist bis zu einer Metastasengröße von zirka 5 cm möglich.
Intrakavitäre Chemotherapie: Die Instillation einer zytostatischen Chemotherapie in ein Hohlorgan oder in eine Körperhöhle wird als intrakavitäre oder als topische Chemotherapie bezeichnet. Beispiele sind die Instillation einer zytostatisch wirksamen Substanz in die Bauchhöhle nach Aszitesdrainage oder in die Pleurahöhle bei Pleuritis carcinomatosa nach Entlastung eines Pleuraergusses. Auch Instillationstherapien in die Harnblase finden Anwendung. Prinzipiell ist das zytotoxische Potenzial der intrakavitär verabreichten Substanzen jedoch als gering anzusehen. Insbesondere bei der Instillation in die Bauchhöhle oder die Pleurahöhle erfolgt eine rasche Resorption des Zytostatikums, sodass eine tumorizide Konzentration meist nur kurzzeitig aufrechterhalten werden kann. Somit werden intrakavitäre Therapien heute nicht mehr mit der Vorstellung durchgeführt, eine lokale Zytotoxizität zu erreichen.
Sinn einer Zytostatikaapplikation in die Pleurahöhle oder in den Perikardbeutel ist, eine Verklebung der Pleura-, respektive Perikardblätter zu erzielen (Pleurodese oder Perikardiodese). Die intrakavitäre Chemotherapie bei einer Peritonealkarzinose ist nahezu gänzlich verlassen worden und hat keinen prognostischen Wert. Bei intrakavitären Therapien im Bereich der Harnblase (bei der Behandlung von Urothelkarzinomen) liegt der Instillation neben dem zytotoxischen Effekt zugrunde, dass durch die Instillation eine immunologische Reaktion ausgelöst wird, die sich gegen das Malignom richtet. So ist es bei der Behandlung des Blasenkarzinoms aus prognostischer Sicht unerheblich, ob das Zytostatikum Mitomycin oder das Immunstimulans Bacille-Calmette-Guérin (BCG) intravesikal appliziert wird (siehe auch Teil Durchführung Abschnitt Lokoregionale Applikation).
Pleurodese: Eine Pleurodese ist bei rezidivierenden Pleuraergüssen, die sich durch zytostatische Therapie der Grundkrankheit nicht bessern, indiziert. Voraussetzung ist die vollständige Entleerung des Pleuraergusses mittels Saugdrainage, anschließend erfolgt die Instillation eines Tetrazyklins oder Zytostatikums, wie Bleomycin oder Mitoxantron. Dadurch wird eine Entzündungsreaktion ausgelöst, durch die beide Pleurablätter miteinander verkleben. Darüber hinaus kann eine Pleurodese thorakoskopisch erfolgen, wobei dann zur Instillation Talkumpuder verwendet wird. Nach Einbringen der entsprechenden Substanz muss sich der Patient mehrfach in etwa 30-minütigen Abständen in eine andere Lageposition bringen (drehen von der linken Seite auf den Rücken und dann auf die rechte Seite usw.), um das Instillat im Pleuraraum zu verteilen. Eine konventionelle Pleurodese ist in etwa 60–70 % der Fälle erfolgreich, eine thorakoskopische Pleurodese in bis zu 90 %. Eine erfolgreiche Pleurodese ist für den Patienten initial schmerzhaft (aufgrund der Entzündungsreaktion im Pleuraspalt) und erfordert eine analgetische Therapie mit Tramadol oder Opiaten, nicht jedoch mit nichtsteroidalen Entzündungshemmern wie Ibuprofen.
Perikardiodese: Eine Perikardiodese wird zur Behandlung rezidivierender Perikardergüsse durchgeführt. Voraussetzung ist auch hier die Entleerung mittels Perikardpunktion, die nur durch einen geübten Arzt erfolgen sollte (in der Regel Kardiologe). Anschließend erfolgt die Instillation der Reizsubstanz. Perikardiodesen sind deutlich seltener erforderlich als Pleurodesen. Bei erfolglosen Perikardiodesen kann als weitere Möglichkeit der Ergussbehandlung eine thoraxchirurgische Perikardfensterung erwogen werden.
2.3.6 Dosisintensivierte Chemotherapie und Hochdosis-Chemotherapie mit Stammzellunterstützung
Jürgen Barth
Moderne Protokolle von Kombinations-Chemotherapie-Schemata induzierten zum Teil beeindruckende Tumorrückbildungen bis hin zu (klinisch) kompletten Remissionen. Leider kommt es oftmals zu Rezidiven mit sogenannter sekundärer Chemotherapieresistenz gegen die anfangs erfolgreich eingesetzten Substanzen. Wenn die Tumorzellen auch zunächst sensibel auf die Zytostatika reagieren, so scheinen diese unter konventionellen Dosierungen nicht in der Lage zu sein, den malignen Zellklon vollständig abzutöten. Unter diesen Bedingungen sind einer Dosissteigerung (Eskalation) der wirksamen Substanzen durch therapiebedingte Nebenwirkungen (Organtoxizität) enge Grenzen gesetzt. Zu den am häufigsten beobachteten Nebenwirkungen gehört die Suppression der Blutbildung (Hämatopoese). Ist diese Myelotoxizität die dosislimitierende Nebenwirkung einer (Kombinations-)Chemotherapie, so lassen sich mittlerweile dennoch Zytostatikadosierungen eskalieren, da diese Nebenwirkung durch Retransfusion peripherer Blutstammzellen beherrschbar ist. Weiterhin lässt sich die sogenannte Dosisintensität – definiert als Dosis pro Zeiteinheit, ausgedrückt in mg/m2 pro Woche, ungeachtet des Schemas, der zeitlichen Abfolge oder des Applikationsweges – durch eine Verkürzung des Dosierungsintervalls erhöhen. Beide Ansätze sollen bei chemosensiblen Tumoren zu einem erhöhten Absterben von Tumorzellen führen. Damit diese Behandlungskonzepte funktionieren, müssen – theoretisch – bestimmte Bedingungen vorgegeben sein.
Seitens des Tumors wäre ideal, wenn er folgende Eigenschaften aufweisen würde:
- exponentielles Tumorwachstum
- konstante Wachstumsfraktion
- konstante Verdopplungszeit des Tumors
- keine Zellfraktion in der G0-Phase (keine schlafenden Zellen; siehe Abschnitt Wirkungs- und Resistenzmechanismen)
- keine Resistenzen der Tumorzellen
- Sensibilität der Tumorzellen auf die applizierten Zytostatika
Die eingesetzten Zytostatika sollten folgende Idealeigenschaften zeigen:
- Abtötungskinetik erster Ordnung bei adäquater Dosierung (je höher die Dosierung, umso höher die Zellabtötungsrate)
- Einzelsubstanz ist bereits in konventioneller Dosierung wirksam
- im höheren Dosisbereich weist sie eine möglichst steile und kontinuierliche Dosis-Wirkungs-Kurve auf (je höher die Dosis, umso stärker die Tumorzellabtötung)
- ihr Einsatz, allein oder in Kombination, ist nicht durch andere Organtoxizitäten limitiert, außer durch Hämatotoxizität
Eine Dosiseskalation ist dann möglich, wenn die Hämatotoxizität durch Gabe hämatopoetischer Wachstumsfaktoren (G-CSF, GM-CSF) in Kombination mit Separation und Retransfusion peripherer Blutstammzellen (weitestgehend) beherrscht werden kann. Mittlerweile hat sich der Ausdruck Stammzelltransplantation im Sprachgebrauch durchgesetzt (peripheral blood stem cell transplantation = PBSCT). Eine deutliche Dosissteigerung ist insbesondere bei folgenden Substanzen möglich:
- Alkylanzien allgemein (inklusive Carboplatin)
- Epipodophyllotoxin-Derivate
- Mitoxantron/Epirubicin
- Taxane
2.4 Stammzelltransplantation (Blut/Knochenmark)
2.4.1 Periphere Stammzelltransplantation (PBSCT, autolog/allogen)
Nach Applikation hämatopoetischer Wachstumsfaktoren, auch koloniestimulierende Faktoren (colony stimulating factors = CSF) genannt, aber auch nach einer myelosuppressiven Chemotherapie als Proliferationsreiz an sich, werden mit den vollständig ausgereiften Granulozyten/Makrophagen noch weitere Blutzellen verschiedener Reifungsstadien verstärkt in das periphere Blut ausgeschwemmt. In der sogenannten CD34-positiven Zellfraktion (CD34 ist ein immunologischer Oberflächenmarker von Zellen; CD = cluster of differentiation oder cluster of determinants) befinden sich offenbar auch „echte Blutstammzellen“. Die Mobilisation von CD34-positiven Zellen wird schon durch Zytokine (CSF) allein beträchtlich erhöht. Eine Chemotherapie ist ein zusätzlicher „Reiz“ zur Aktivierung und Mobilisierung von Stammzellen. Eine Kombination von Wachstumsfaktoren und Chemotherapie bringt deutlich höhere Leukaphereseausbeuten (ein auf Zentrifugation basierendes Separationsverfahren) an peripheren Stammzellen als jede Methode für sich allein (alleinige Verabreichung von Zytostatika oder Zytokinen (G-CSF, GM-CSF)). Eine kombinierte zytotoxische/zytokininduzierte (Chemotherapie + G-CSF oder GM-CSF) Mobilisation ist mittlerweile üblich. Die initiale Medikation wird als (Hochdosis-)Mobilisierungsschema bezeichnet. Der Mobilisation folgt eine an mehreren Tagen mit Zellseparation durchgeführte Apheresephase für CD34-positive Zellen (Abbildung rechts).
Das Stammzellkonzentrat wird nach Zugabe von Dimethylsulfoxid (= DMSO; dient als „Gefrierschutz“, damit die Zellen beim Einfrieren nicht platzen und vital bleiben) in flüssigem Stickstoff tiefgefroren. Diese Zellkonzentrate stehen den Patienten als autologe Stammzelltransplantate zur Unterstützung nach der später durchgeführten Hochdosis-Chemotherapie in der stark myelosuppressiven Phase zur Verfügung. Sowohl Mobilisations- als auch Hochdosis-Schema (HD-CX) sind üblicherweise tumorspezifisch.
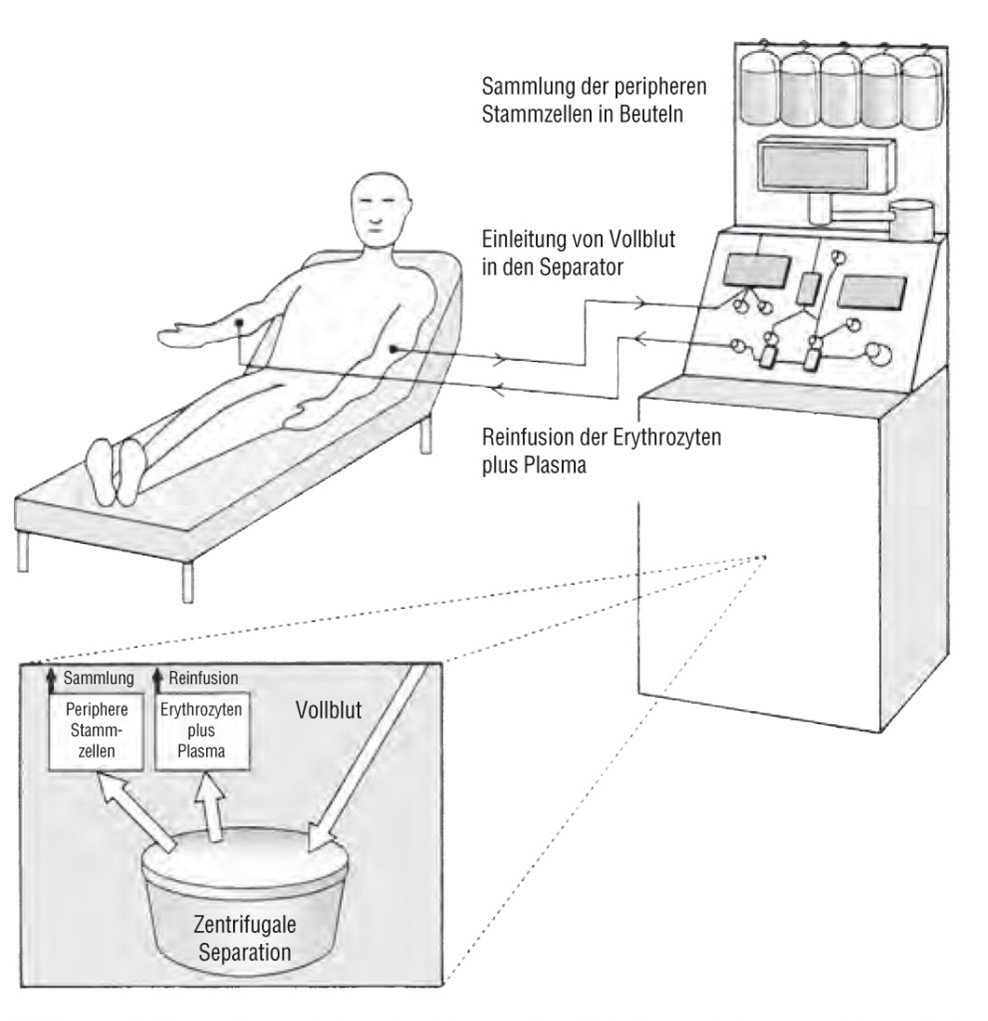
Weiterhin wird die periphere Stammzelltransplantation (PBSCT) als Alternative zur autologen Knochenmarktransplantation (Knochenmark vom Patienten selbst) verwendet. Im Vergleich zur Knochenmarktransplantation ist die periphere Stammzelltransplantation weniger belastend für den Patienten. Sie kann auch bei Patienten durchgeführt werden, bei denen eine Vollnarkose (zur Knochenmarkpunktion) kontraindiziert ist. Auch chemotherapeutisch vorbehandelte Patienten oder solche mit bestrahlten Beckenknochen können behandelt werden. Ebenso Patienten mit einem für eine autologe Knochenmarktransplantation ungeeigneten, zum Beispiel noch funktionstüchtigen, aber hypozellulären Knochenmark.
Mittlerweile verdrängt allmählich die allogene (vom Fremdspender) PBSCT die allogene Knochenmarktransplantation (KMT). Es konnte gezeigt werden, dass durch eine Applikation von G-CSF bei gesunden Probanden die Zahl der im peripheren Blut auftretenden Stammzellen in einem solchen Ausmaß erhöht werden kann, dass sogar eine Zytapherese mit suffizienten Ausbeuten an CD34-positiven Zellen möglich ist. Auch für den Fremdspender ist eine allogene periphere Stammzelltransplantation weniger belastend und mit weniger Aufwand durchzuführen. Es wird nur eine Probepunktion im Beckenbereich vorgenommen. Eine Vollnarkose und eine Hospitalisierung sind nicht notwendig. Es gibt kaum einen Nachbeobachtungsaufwand.
Welche Tumoren sind für dieses Verfahren geeignet?
- Tumoren mit einem prognostisch ungünstigen Erkrankungsstadium bei potenziell kurativer Therapie-Intention
- Tumoren mit Chemosensitivität gegenüber den eingesetzten Zytostatika
- geringes Tumorvolumen (z. B. Plasmozytom, akute Leukämien, hochmaligne Non-Hodgkin-Lymphome oder Morbus Hodgkin)
Welche Patienten können diesem Verfahren unterzogen werden?
- Patienten mit gutem Allgemeinzustand
- Patienten mit guter Nieren- und Leberfunktion
- Patienten mit prätherapeutisch ausreichenden Mengen an separierten peripheren Stammzellen
- Patienten, die eine möglichst geringe Tumorzellkontamination im autologen Separat haben
Eine HD-CX ist wirksam beim Plasmozytom, rezidivierenden Lymphomen (Hodgkin, Non-Hodgkin), kindlichen Sarkomen und Neuroblastomen, adulten Ewing-Sarkomen, Ovarial- und Keimzelltumoren. Ergebnisse kontrollierter Studien – insbesondere beim Mammakarzinom – stehen aber noch aus.
2.4.2 Knochenmarktransplantation
Norbert Schleucher
Unter einer Knochenmarktransplantation (KMT) versteht man die therapeutische Übertragung eines Spenderknochenmarks. Dabei handelt es sich wie bei der Leber- und Nierentransplantation um eine Organtransplantation. Das transplantierte Organ ist das Knochenmark. Wie bei jeder Organtransplantation sind Behandlungen mit Immunsuppressiva erforderlich. Das bedeutet, dass man bei einer KMT mit einem Nebenwirkungsspektrum wie bei einer Organtransplantation rechnen muss. Da jedoch das gesamte blutbildende System und das Immunsystem ersetzt werden, resultieren auch spezifische Komplikationen (siehe Abschnitt Komplikationen einer Knochenmarktransplantation). Prinzipiell handelt es sich bei der Knochenmarktransplantation um das risikoreichste Therapieverfahren im Bereich der Hämatologie und internistischen Onkologie.
2.4.2.1 Indikationen zur Knochenmarktransplantation
Knochenmarktransplantationen werden bei Leukämien und hochmalignen Lymphomen durchgeführt. Insbesondere im Bereich der akuten Leukämien konnte die KMT eine entscheidende Prognoseverbesserung erbringen. Aber auch bei chronischen Leukämien, insbesondere der chronischen myeloischen Leukämie, kann die KMT möglicherweise einen kurativen Ansatz bedeuten. In früheren Zeiten wurde die Knochenmarktransplantation auch bei Hodenkarzinomen durchgeführt, hier wurde sie jedoch zugunsten der nebenwirkungsärmeren Stammzelltransplantation verlassen.
Einen festen Platz hat die Knochenmarktransplantation bei der Therapie der akuten myeloischen und der akuten lymphatischen Leukämie. Hier wird sie als Konsolidierung nach Erreichen einer kompletten Remission durch eine konventionelle zytostatische Chemotherapie eingesetzt. Dabei orientiert sich die Indikationsstellung zur KMT an der Risikokonstellation des Patienten, insbesondere definiert über seine chromosomalen Aberrationen. Als Faustregel erfolgt jedoch der Einsatz der Knochenmarktransplantation bei der akuten myeloischen Leukämie in der ersten kompletten Remission, bei der akuten lymphatischen Leukämie (ohne Vorliegen einer Hochrisikokonstellation) in der zweiten kompletten Remission (also nach Erreichen einer kompletten Remission im Rahmen einer Rezidivtherapie).
2.4.2.2 Voraussetzungen für eine Knochenmarktransplantation
Unabdingbare Voraussetzung für eine Knochenmarktransplantation ist die HLA-Kompatibilität von Knochenmarkspender und Knochenmarkempfänger. Das heißt, die humanen Leukozytenantigene von Spender und Empfänger müssen übereinstimmen. Dabei wird jedoch ein nicht übereinstimmendes Merkmal (mismatch) akzeptiert. Die Blutgruppen von Spender und Empfänger müssen für eine KMT nicht übereinstimmen; in diesen Fällen zeigt der Knochenmarkempfänger nach der Transplantation meistens die Blutgruppe des Spenders. Darüber hinaus dürfen beim Knochenmarkempfänger keine gravierenden Grunderkrankungen seitens des Herzens oder der Lunge bestehen. Die Knochenmarkempfänger dürfen in der Regel das 60. Lebensjahr nicht überschritten haben, weil ab diesem Alter die therapieinduzierte Morbidität und Mortalität deutlich ansteigen. Beim Knochenmarkspender müssen – abgesehen von Tumorfreiheit – keine weiteren Voraussetzungen erfüllt werden. Weitere Voraussetzung direkt vor Durchführung der Knochenmarktransplantation mit Immunsuppression ist die Infektfreiheit des Knochenmarkempfängers, da Infektionen unter Immunsuppression generalisieren und septikämische Verläufe nehmen können.
2.4.2.3 Praktische Durchführung der Knochenmarktransplantation
Grundlegend wird zwischen einem Familienspender und einem Fremdspender unterschieden. Als Familienspender gelten dabei Eltern und Geschwister des Patienten, aber auch Verwandte zweiten Grades. Demgegenüber werden alle nicht familiären Knochenmarkspender als Fremdspender bezeichnet. Passende Fremdspender werden über Spenderkarteien ermittelt. Diese Spenderdatenbanken sind sehr umfangreich, sodass aktuell die Wahrscheinlichkeit, einen passenden Fremdspender zu finden, bei etwa 80 % anzusiedeln ist.
Bei einer Knochenmarkspende erfolgt die Entnahme des Knochenmarks aus beiden Beckenkämmen des Spenders. Weil es sich dabei um einen größeren und schmerzhaften Eingriff handelt, ist zwar eine Vollnarkose erforderlich, aber die Knochenmarkspende an sich ist mit keinem nennenswerten Risiko verbunden. Nach einer entsprechenden Aufbereitung erfolgt dann die Übertragung des fremden Knochenmarks in Form einer intravenösen Infusion. Das infundierte Spenderknochenmark erreicht auf dem Blutweg die Knochenmarkräume und siedelt sich dort an.
2.4.2.4 Konditionierungstherapie
Bevor sich das transplantierte Knochenmark in Knochenmarkräumen ansiedeln kann, ist die „Zerstörung“ des wirtseigenen Knochenmarks eine unabdingbare Voraussetzung. Diese „Zerstörung“ des wirtseigenen Knochenmarks wird als Konditionierungstherapie bezeichnet. Dabei werden hohe Zytostatikadosen eingesetzt, meist hochdosiertes Cyclophosphamid. Zusätzlich wird in den meisten Fällen eine Ganzkörperbestrahlung durchgeführt. Aufgrund der hohen Dosisintensität beider Modalitäten wird das wirtseigene Knochenmark vernichtet.
2.4.2.5 Komplikationen einer Knochenmarktransplantation
Erhöhte Infektanfälligkeit: Wie jede Organtransplantation erfordert auch eine KMT eine Immunsuppression, um eine lymphozytäre Immunantwort gegen das Spenderknochenmark auszuschließen. Diese Immunsuppression erfolgt mit Kortikosteroiden (z. B. Prednison) in Kombination mit Cyclosporin A (Sandimmun®). Neuere Immunsuppressiva sind die Substanz Tacrolimus (Prograf®) oder Mycophenolat mofetil (Cell Cept®). Diese Immunsuppression bedingt eine erhöhte Infektanfälligkeit des Knochenmarkempfängers für bakterielle oder virale Infektionen, aber auch für opportunistische Erreger. Bei immunsupprimierten Patienten besteht bei jeder Infektion das Risiko einer Sepsis mit Todesfolge.
2.4.2.5.1 Abstoßungsreaktion
Eine weitere Komplikation, wie bei jeder Organtransplantation, ist das Auftreten einer Abstoßungsreaktion, das heißt, verbliebene wirtseigene Knochenmarkzellen, insbesondere Lymphozyten, richten sich gegen das empfangene Knochenmark. Im Zuge einer Knochenmarktransplantation wird diese Reaktionsform des Organismus auch als „host versus graft reaction“ bezeichnet. Die umgekehrte Reaktion nennt man „graft versus host reaction“. Hierbei reagieren Lymphozyten des empfangenen Knochenmarks gegen wirtseigene Zellen und induzieren eine immunogen bedingte Zytolyse gesunder Wirtszellen. Diese Reaktionsform ist insbesondere wahrscheinlich, wenn nicht exakt HLA-identisches Knochenmark transplantiert worden ist (das heißt beim Auftreten von mismatches). Stammt das gespendete Knochenmark demgegenüber von einem HLA-identischen Spender (zum Beispiel eineiiger Zwilling), so ist eine „graft versus host reaction“ nahezu ausgeschlossen. Die Graft-versus-Host(GvH)-Krankheit kann dabei akut oder chronisch auftreten. Sie kann alle Organe des Empfängers betreffen.
Häufig manifestiert sich die Graft-versus-Host-Krankheit an der Haut. Hier sieht man bei der chronischen GvH-Erkrankung bräunliche Verfärbungen der gesamten Haut, die dann vulnerabler ist als gesunde Haut. Eine GvH-Reaktion des Darmes führt zu Durchfällen. Auch tritt im Rahmen von akuten GvH-Reaktionen meistens Fieber auf. Als Vorteil einer GvH-Reaktion ist es zu sehen, dass sich das Spenderknochenmark nicht nur gegen wirtseigene Zellen richtet, sondern auch gegen erkrankte Zellen, die sich noch im Wirtsorganismus befinden. Dies wird als Graft-versus-Disease-Phänomen bezeichnet. Diese GvD-Reaktion kann in bestimmten Fällen therapeutisch ausgenutzt werden. So kann man bei Frührezidiven einer Leukämie nach Knochenmarktransplantation durch Absetzen der Immunsuppressiva eine Graft-versus-Host-Krankheit und damit auch eine Graft-versus-Disease-Reaktion auslösen.
Letalitätsrisiko: Jede Knochenmarktransplantation ist mit einem potenziellen Letalitätsrisiko behaftet. Dies wird in der gängigen Literatur mit 5–10 % beziffert. Das Letalitätsrisiko einer Knochenmarktransplantation steigt mit dem Alter des Patienten an. So beträgt das Letalitätsrisiko bei 65-jährigen Patienten beispielsweise 25–30 %!
2.4.2.6 Minitransplantation (allogen)
Als Minitransplantation wird die Übertragung nicht des gesamten Spenderknochenmarks, sondern von Fremdspenderstammzellen bezeichnet. Dabei erfolgt nach Gewinnung des Knochenmarks ein spezielles Aufbereitungsverfahren, sodass nur CD34-positive Stammzellen reinfundiert werden. Es handelt sich somit um eine allogene Stammzelltransplantation im Gegensatz zur autologen Stammzelltransplantation, bei der eigene Stammzellen des Patienten retransfundiert werden. Die Minitransplantation ist komplikationsärmer als eine Knochenmarktransplantation. Heute gilt die Minitransplantation in verschiedenen Indikationen der allogenen Knochenmarktransplantation als gleichwertig und ist als ein Standardverfahren zu betrachten.
2.4.2.7 Prophylaxen im Rahmen der Knochenmarktransplantation
Da im Rahmen der Konditionierungstherapie das wirtseigene Immunsystem komplett zerstört worden ist und die Ausreifung des transplantierten Knochenmarks bis hin zum funktionsfähigen Immunsystem eine gewisse Zeitspanne benötigt, ist der Patient während dieser Zeit (zirka 3–6 Wochen) einer hohen Infektgefährdung ausgesetzt. Der Knochenmarkempfänger ist in dieser Zeit ohne Immunsystem. Aus diesem Grunde hat die Infektprophylaxe bei Knochenmarkempfängern höchste Priorität. Im klinischen Alltag ist daher eine Umkehrisolation des Patienten Pflicht.
Bei dieser Umkehrisolation befindet sich der Patient in einem Isolationszimmer, das nur durch eine Schleuse betreten werden kann. In dieser Schleuse müssen Arzt und Pflegepersonal eine entsprechende Schutzkleidung anlegen, um den immunsupprimierten Patienten vor Keimen zu schützen. Diese Schutzkleidung umfasst eine vollständige Abdeckung des Kopfes, einen Mundschutz, Handschuhe und keimarme Kittel sowie keimarme Schuhüberzüge. Eine gründliche Händedesinfektion ist erforderlich. Der Patient in der Umkehrisolation darf sein Zimmer nicht verlassen. Sollte dies aus medizinischen Gründen zwingend erforderlich sein, so muss der Patient Handschuhe und einen speziellen Mundschutz tragen. Dieser Mundschutz enthält ein Filtersystem, das aerogen übertragbare Keime mit einer bestimmten Porengröße herausfiltert. Darüber hinaus sind sogar die Mahlzeiten eines knochenmarktransplantierten Patienten zu sterilisieren. Geeignete bzw. ungeeignete Mahlzeiten für Patienten in Umkehrisolation siehe Tabelle unterhalb.
Die Räumlichkeiten werden auch als „life islands“ bezeichnet. Besucher dürfen trotz aller Vorsichtsmaßnahmen das Zimmer eines solchen Patienten nicht betreten, es sind lediglich Kontakte aus einem Besucherraum her erlaubt, der durch eine Glaswand vom Patientenzimmer abgetrennt ist. Die Kommunikation muss über Telefonanlagen erfolgen.
Aus diesen Schilderungen wird ersichtlich, dass eine Knochenmarktransplantation für den Patienten eine hohe physische, aber auch eine hohe psychische Belastung darstellt.
| Lebensmittel | Geeignet | Ungeeignet |
| Fleisch/Geflügel | gut durchgegart (kochen, dünsten, dämpfen, grillen, braten) | rohes/halbrohes Fleisch (Mett, Tartar, Roastbeef) |
| Fisch | gut durchgegart | roher Fisch, Fischsalate, Schalen- und Krustentiere |
| Wurst/Brotbelag | Brüh- und Kochwurst | Rohwurstsorten (Salami, Mettwurst …), Pflanzenpastete |
| Obst | Kompott, Schälobst (Orange, Banane, Mandarine) | Trockenobst, Obstsalate, Nüsse, Mandeln, nicht schälbares Obst, wie Trauben, Pfirsiche, Kirschen, Kernobst (Apfel, Birne) |
| Käse | Schmelzkäseportionen, wärmebehandelter Frischkäse in Portionen | Schnittkäse, Hartkäse, Edelpilzkäse, Weichkäse, Schafs- und Ziegenkäse |
| Milch und Milchprodukte | Kondensmilch, sterilisierte Milch, Pudding und Brei frisch gekocht | Rohmilch, Vorzugsmilch, pasteurisierte Milch, H-Milch, Eis, Quark, Joghurt, Quark- und Joghurtspeisen |
| Eier | nur gekocht oder gebraten | roh, weichgekocht, Mayonnaise |
| Getreideerzeugnisse | nur gegart | Getreidekörner, Kleie, Müslimischungen, Haferflocken, Getreideflocken |
| Brot und Backwaren | alle frischen Brotsorten | Brot älter als 1 Tag, Schnittbrot |
| Gewürze | gegart (mitgekocht) | Kräuter (frisch oder getrocknet); kalte Soßen, rohe Zwiebeln, Knoblauch, nicht nachwürzen nach dem Kochen! |
| Getränke | alle kohlensäurehaltigen Getränke wie Cola, Limo, Mineralwasser, frisch gekochter Tee oder Kaffee, Fruchtsäfte als Portionsware | kohlensäurefreies Mineralwasser, länger offen stehende Getränke (Mineralwasser, Cola, Limo, Fruchtsäfte, Tee …) |
| Süßigkeiten | Schokolade ohne Mandeln und Nüsse, Gummibärchen, Bonbons, Kuchen und Gebäck ohne Mandeln und Nüsse | Traubennussschokolade, Schokolade mit Mandeln und Nüssen, Kuchen und Gebäck mit Nüssen, Mandeln, Rosinen |
2.5 Hyperthermiebehandlung
Unter einer Hyperthermie versteht man die iatrogene Erwärmung bestimmter Körperregionen oder des ganzen Körpers auf eine Zieltemperatur von 40–42 °C. Im Rahmen der antineoplastischen Therapie wird eine Hyperthermie meist in Kombination mit einer zytostatischen Chemotherapie eingesetzt.
Bei einer Ganzkörper-Hyperthermie erfolgt dabei die Wärmezufuhr zum Beispiel durch Wassermatten oder eine Bluterwärmung mittels eines extrakorporalen Kreislaufsystems.
Bei organbegrenzter lokoregionärer Hyperthermie erfolgt die Wärmeerzeugung durch Ultraschall- oder elektromagnetische Wellen.
Der Hyperthermiebehandlung liegen verschiedene Wirkprinzipien zugrunde. So kann eine Temperatur von 42 °C zur Denaturierung von Tumorproteinen und damit zum Zelltod von Tumorzellen führen. Darüber hinaus wird die Hyperthermie in Kombination mit Chemotherapie eingesetzt unter der Vorstellung, dass durch die Hyperthermie die Barriere der Tumorzellen für die Zytostatika „gelockerter und durchlässiger“ wird.
Eine Hyperthermiebehandlung ist gut möglich bei Tumoren, die an den Extremitäten lokalisiert sind. Hier kann eine isolierte Erwärmung der befallenen Extremität durchgeführt werden. Als Beispiele gelten die hypertherme Extremitätenperfusion mit Melphalan bei der Behandlung maligner Melanome oder die arterielle Zytostatikaperfusion in Hyperthermie bei Weichteilsarkomen. Auch bei lokal rezidivierten inoperablen Rektumkarzinomen kann eine Hyperthermie des kleinen Beckens in Kombination mit Chemotherapie indiziert sein. Bei viszeralen Metastasen ist eine Hyperthermiebehandlung schwieriger und technisch aufwendiger. Hier ist Voraussetzung, dass sich der zu erwärmende tumortragende Bezirk nicht in der Nähe großer Gefäße befindet, weil diese Gefäße einen Abtransport der Wärme bedingen. Ebenfalls aufwendig ist die hypertherme intraperitoneale Chemoperfusion (HIPEC) zur Behandlung einer Peritonealkarzinose. Dabei werden Zytostatikalösungen auf 42 °C erwärmt und für 30–90 Minuten im Abdominalraum verteilt.
Insgesamt gilt, dass die Hyperthermiebehandlung risikoreicher wird, wenn innere Organe erwärmt werden, wobei das Behandlungsrisiko mit der Größe des erwärmten Areals steigt. Eine Ganzkörper-Hyperthermie kann potenziell lebensbedrohlich sein.
2.6 Intensivmedizin und Dialyse
In der hämatologischen und onkologischen Routinetherapie werden intensivmedizinische Maßnahmen selten benötigt. So gibt es kein hämatologisches oder onkologisches Therapieverfahren, das zwingend auf einer Intensivstation durchgeführt werden müsste. Autologe Stammzelltransplantationen können in normalen Krankenzimmern durchgeführt werden. Eine Monitor- oder Kreislaufüberwachung ist nicht zwingend erforderlich. Auch neutropenische Patienten mit erhöhter Infektgefährdung müssen nicht auf einer Wachstation behandelt werden.
In bestimmten Situationen kann jedoch – wie in der allgemeinen Inneren Medizin auch – eine respiratorische oder kardiozirkulatorische Überwachung erforderlich werden. Eine intensivmedizinische Behandlung ist nicht zu umgehen, wenn invasive Behandlungsverfahren erforderlich werden. Dazu gehört beispielsweise ein arterielles Monitoring, eine Behandlung mit der CPAP-Maske (continuous positive airway pressure) oder eine Beatmungstherapie.
Indikationen zur intensivmedizinischen Überwachung/Behandlung sind beispielsweise:
- arterielles Monitoring beim septischen Patienten
- hochgradig instabiles Blutdruckverhalten
- höhergradige supraventrikuläre oder ventrikuläre Arrhythmien
- Respiratorische Überwachung bei Pneumoniepatienten
- ausgeprägtes Tumorlyse-Syndrom
- schwere Verlaufsform einer Verbrauchskoagulopathie
- Beatmungstherapie bei respiratorischer Insuffizienz
- Überwachung bei schwerwiegender allergischer Reaktion oder anaphylaktischem Schock (bei Katecholaminpflichtigkeit)
- Kreislaufinstabilität mit Katecholaminpflichtigkeit
- ausgeprägte Koronarspasmen unter 5-FU-Behandlung, die dem Bild einer instabilen Angina pectoris entsprechen
- schwerwiegende Infektionserkrankungen, wie Meningitiden oder Endokarditiden
- drohendes Multiorganversagen
Äußerst wichtig ist es, den invasiven Charakter intensivmedizinischer Maßnahmen nicht zu fürchten und die erforderlichen Maßnahmen ohne zeitliche Verzögerung einzuleiten. So sollte eine notwendige Intubation eines neutropenischen Patienten mit Pneumonie nicht hinausgeschoben werden, weil man fürchtet, dass es sich um eine langwierige Respiratortherapie handelt. Auch das Wissen um die schlechte Prognose eines beatmeten neutropenischen Pneumoniepatienten sollte nicht vor einer maschinellen Beatmung zurückschrecken lassen. Die Beatmungspflichtigkeit resultiert aus der Schwere der Grunderkrankung oder der Schwere der eingetretenen Komplikation und nicht durch die Beatmungsmaschine an sich! Dabei sollte man sich immer vor Augen halten, dass Beatmungsgeräte nicht süchtig machen und dass Endotrachealtuben keine Krankheit sind! Falls bei einem onkologischen Patienten eine Langzeitbeatmung erforderlich ist, so ist diese aufgrund der Schwere der Grunderkrankung erforderlich.
Als Komplikation bestimmter Zytostatikatherapien kann es zu einem vorübergehenden oder dauerhaften Ausfall der Nierenfunktion kommen. Auch bei onkologischen Grunderkrankungen wie Plasmozytomen oder Amyloidosen ist ein akutes oder chronisches Nierenversagen im Verlauf der Erkrankung denkbar. In diesen Fällen ergibt sich die Indikation zur Dialysebehandlung. Bei einer Dialyse erfolgt eine extrakorporale Entgiftung des Blutes mittels eines Dialysegerätes. Dazu ist ein entsprechend großlumiger Zugang erforderlich. Bei der Akutdialyse handelt es sich in der Regel um einen Shaldon-Katheter, bei einer chronischen Dialyse um einen Cimino-Shunt. Die Reinigung des Blutes von Schadstoffen erfolgt dabei extrakorporal an Membransystemen nach den physikalischen Gesetzen von Osmose und Diffusion. Bei beatmeten Patienten kann die CvvHD (kontinuierliche veno-venöse Hämodialyse) Anwendung finden.
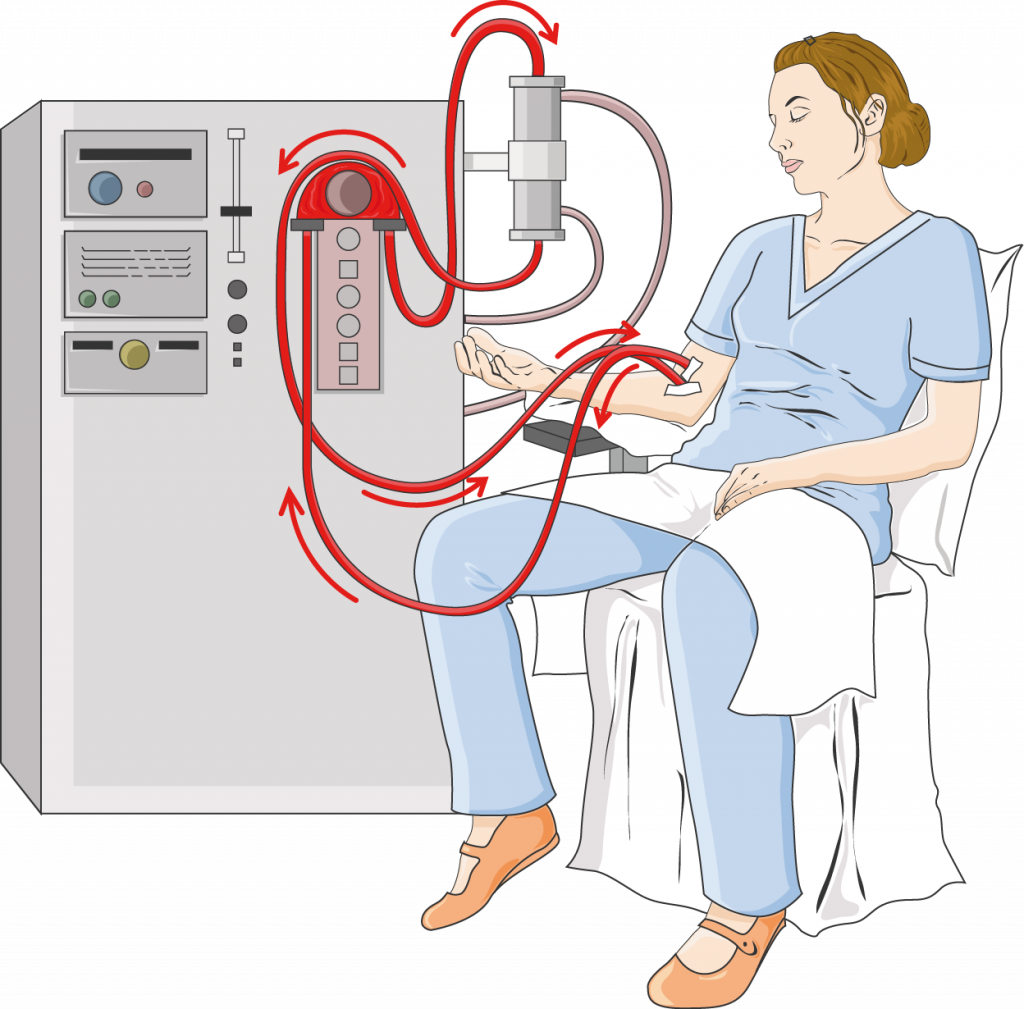
Ein zytostatikainduziertes akutes Nierenversagen ist in den allermeisten Fällen reversibel, eine chronisch fortschreitende zytostatikainduzierte Niereninsuffizienz ist demgegenüber im Terminalstadium dauerhaft dialysepflichtig. Die akute Niereninsuffizienz im Rahmen eines Multiorganversagens, bei Tumorpatienten mit Neutropenie häufig septisches Multiorganversagen, hat eine schlechte Prognose, da neben der Nierenfunktion auch Leber, Lungen- und Kreislaufsystem vom Multiorganversagen betroffen sind.
Bei Patienten mit Plasmozytomen oder Amyloidosen kommt es zur Nierenschädigung im Rahmen der Grunderkrankung durch Ablagerung bestimmter Paraproteine in den Nephronen. Auch hier resultiert eine chronisch fortschreitende Niereninsuffizienz, möglicherweise mit Dialysepflichtigkeit. In einem solchem Fall ist die Indikation zur Nierenersatztherapie gegeben, da bei Ersatz der Nierenfunktion die Patienten durch zeitgleiche zytostatische Chemotherapie mit Kontrolle des Grundleidens noch mehrere Jahre überleben können. Für die Indikation zur Dialysebehandlung gilt wie für die Indikation zur Respiratortherapie, dass die Indikationsstellung nicht aufgrund des malignen Grundleidens allein unterbleiben darf.
Tumorpatienten sind sehr oft auch Schmerzpatienten und wegen der vielerorts nur suboptimal durchgeführten Schmerztherapie in ihrer Lebensqualität deutlich eingeschränkt. Auf die Schmerztherapie wird ausführlich im Teil Schmerz eingegangen.
2.7 Alternative Behandlungsverfahren mit nicht belegter Wirksamkeit
Als „Alternativtherapien“ werden therapeutische Ansätze mit Medikamenten zusammengefasst, die nicht aus dem Bereich der Zytostatika stammen. Der „Alternativtherapien“ bedienen sich keine Onkologen, Chirurgen oder Strahlentherapeuten, sondern Heilpraktiker, Homöopathen, Geistheiler oder Sauerstofftherapeuten. Es handelt sich dabei ausnehmend um Therapieverfahren, die nicht zulasten der gesetzlichen Krankenversicherung verordnet werden dürfen. Sie sind oft letzte Alternative zur Beruhigung von Angehörigen dem Patienten gegenüber oder sind medienwirksam verbreitet.
Die eingesetzten Präparate stammen aus dem Bereich der Homöopathie, sogenannter Drogenpräparate, Phytotherapeutika oder chemisch definierter Alternativtherapeutika. Allen diesen Präparaten und den verschiedenen Substanzklassen gemeinsam ist ein nicht definierter Wirkmechanismus (da Bestandteil einer Heilslehre). Darüber hinaus sind die Indikationsstellungen der Hersteller unklar. So werden viele dieser Medikamente als universelle Tumorpräparate bezeichnet, ohne dass der Biologie verschiedener Malignomerkrankungen Rechnung getragen wird.
Merke: Insgesamt muss von der Anwendung „alternativer Therapien“ in großem Umfang abgeraten werden. Neben den fehlenden Wirkungsbeweisen und der unklaren Indikationsstellung ist auch das Nebenwirkungsspektrum der Alternativtherapeutika in der Regel zu wenig definiert. Sollte ein Patient die Anwendung solcher Therapien wünschen, ist eine vorherige Besprechung mit dem behandelnden Onkologen dringend anzuraten, insbesondere um Wechselwirkungen mit zytostatischen Therapien auszuschließen.
Von Patienten werden Ärzte und Pflegepersonal sehr häufig nach sogenannten Alternativtherapien gefragt. Dies entspricht bei fortgeschrittenen Erkrankungen einem „Klammern an den letzten Strohhalm“. Da viele dieser Präparate sehr teuer sind und nicht zulasten der Krankenversicherungen verordnet werden dürfen, sondern vom Patienten selbst zu finanzieren sind, ist es hier ärztliche und auch pflegerische Aufgabe, Patienten vor finanziellem Schaden bei meist nicht heilbarer Krankheit zu schützen.
Sehr häufig verwendete Präparate sind die sogenannten Mistellektine. Bei Einsatz solcher Präparate handelt es sich um eine Adoption aus dem anthroposophischen in den medizinischen Bereich. So wird der Mistel eine Schwächung der physischen Fremdbildung, des Tumors, durch Entzug seines Wucheräthers und seiner Lebenskraft zugeschrieben. Es wird postuliert, dass die Mistel eine antitumorale Symbiose mit dem Patienten gegen seinen Tumor eingeht. Eine genaue biochemische oder pharmakologische Definition des Wirkmechanismus ist jedoch nicht bekannt. Positive In-vitro-Daten und auch positive randomisierte Studien (verglichen gegen Placebo oder konventionelle Zytostatika) fehlen. Trotzdem werden Mistelpräparate sehr häufig bei Patienten mit kolorektalem Karzinom eingesetzt. Auch hier vermag die Mistel allein den Spontanverlauf der Erkrankung nicht zu beeinflussen. Immerhin ist auch keine Antagonisierung einer klassischen 5-FU-haltigen zytostatischen Chemotherapie bekannt. Manche Patienten beschreiben allerdings eine nicht objektivierbare Verbesserung ihres Allgemeinzustandes unter Mistelinjektionen.
Von Anwendern alternativer Therapieverfahren wird häufig in Medien und Zeitschriftenanzeigen damit geworben, dass einerseits pflanzliche Präparate verwendet werden und dass diese andererseits keine Nebenwirkungen auslösen. In diesem Zusammenhang sei darauf hingewiesen, dass auch die klassischen Zytostatika zu über 50 % pflanzlicher Herkunft sind. So stammen hochwirksame Zytostatika aus Baumblättern, Baumrinden oder niederen Gewächsen. Als sehr modernes Beispiel sei das Zytostatikum Paclitaxel (Taxol®) angeführt, das aus der Rinde der südpazifischen Eibe (Taxus brevifolia) gewonnen wurde und das mittlerweile semisynthetisch hergestellt werden kann.
Alternativmedizinische Präparate sind nicht nebenwirkungsfrei! Entzündungen und Verhärtungen an Mistel-Injektionsstellen gibt es genauso wie schwere allergische Reaktionen nach Einsatz von Thymuspräparaten. Mit der Behandlung dieser Nebenwirkung ist dann der Nicht-Mediziner in aller Regel überfordert.
3. Wirkungs- und Resistenzmechanismen von Antitumortherapeutika
Jürgen Barth
3.1 Wirkungsmechanismen von Antitumortherapeutika
Zytostatika sind Zellgifte mit unterschiedlichen Angriffspunkten innerhalb der Zellen. Die Selektivität auf Tumorzellen ist eine relative, aufgrund der insbesondere bei hoher Tumorlast deutlich höheren Stoffwechselrate der entarteten Zellen. Weiterhin bestimmen auch die physiko-chemischen Eigenschaften die Gewebeverteilung der einzelnen Substanzen. So können beispielsweise nur besonders lipophile Stoffe in therapeutisch relevanten Mengen eine intakte Blut-Hirn-Schranke überwinden. Viele der (klassischen) Zytostatika greifen innerhalb der Zelle in diverse, nicht zwingend tumorspezifische Stoffwechselvorgänge während des Zellzyklus ein. Die diesbezüglich wesentlichen Grundlagen werden hier in aller Kürze zusammengefasst.
Der Zellzyklus – der Lebenszyklus der Zelle mit der Abfolge der Zellreifung und -teilung – kann in die fünf Phasen G1, S, G2, G0 und M eingeteilt werden (G steht für „gap“ = Lücke, zeitliche Lücken zwischen der DNS-Verdopplung; S steht entsprechend für Synthese der DNS). Vorgänge innerhalb dieser Phasen sind:
G1: RNS-Synthese
S: DNS-Verdoppelung
G2: Intervall zwischen Mitose und DNS-Verdoppelung
G0: Ruhephase von ansonsten teilungsaktiven Zellen. Der eigentliche Zellzyklus ist durchlaufen („schlafende Zellen“). Chemotherapeutika können in dieser Phase kaum Zellschäden herbeiführen. In dieser Ruhephase können durch Zytostatika verursachte Schäden repariert werden, bevor es zu letalen Folgen während oder nach der eigentlichen Zellteilung kommt.
M: Mitose: Kernteilungsvorgang, bei dem aus einem Zellkern zwei Tochterkerne gebildet werden. Eine weitere Unterteilung des zeitlichen Ablaufs in Pro-, Meta-, Ana- und Telophase ist möglich.
Die einzelnen Zellzyklusphasen sind in der nachfolgenden Abbildung schematisch verdeutlicht. Bezogen auf den Zellzyklus können Zytostatika in phasenspezifische (z. B. Antimetaboliten) und in phasenunspezifische (z. B. Alkylanzien) Wirkstoffe eingeteilt werden.
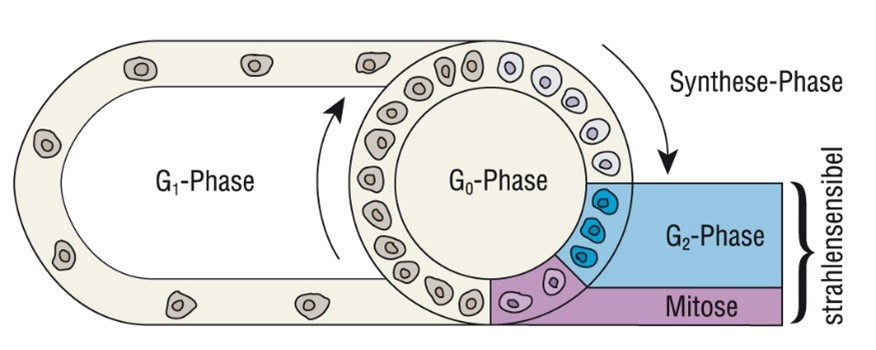
Die DNS ist als zentraler Informationsspeicher für die Funktionen innerhalb der Zelle und des Zellverbandes oftmals (aber nicht ausschließlich) primäres Angriffsziel von Chemotherapeutika. Die DNS ist eine Nukleinsäure mit der allgemeinen Zusammensetzung (Base-[2-Desoxyribose]-Phosphorsäure), wie in der folgenden Abbildung dargestellt. Die sogenannten Basen sind die Purine Adenin und Guanin sowie die Pyrimidine Thymin und Cytosin. Diese sind jeweils durch 3’,5’-Desoxyribosephosphorsäurediester-Brücken miteinander verbunden.
Das DNS-Molekül besteht – bezüglich der Basen – aus zwei komplementären Strängen, die über Wasserstoffbrücken eine Doppelhelix bilden (Abbildung unterhalb). Es paaren sich immer die komplementären Basen Adenin mit Guanin sowie Thymin mit Cytosin. Die Basenfolge des einen Stranges bestimmt damit die Sequenz des anderen. In der Reihenfolge der Basen ist die jeweilige genetische Information kodiert.
Die DNS stellt den genetischen Kode für die Eiweißbiosynthese (Struktur- und Enzymproteine) und die Erbmerkmale dar. Geschädigte DNS kann durch zelleigene Enzymsysteme repariert werden. Für eine Defektentfernung werden Nukleasen, für eine Neusynthese von Segmenten DNS-Polymerasen aktiviert. Der Einbau einer neuen oder korrigierten Basensequenz erfolgt durch Ligasen. Werden für Genprodukte Informationen aus der DNS benötigt oder soll sie für eine Zellteilung dupliziert werden, muss sie, um abgelesen werden zu können, entspiralisiert, aufgeschnitten und nach Ablesen der benötigten Informationen wieder verknüpft und verdrillt werden. Dabei wird die DNS in einen komplexen Zwischenzustand überführt, der Replikationsgabel genannt wird. Die Aufgabe, diesen Zwischenzustand zu erzeugen, übernehmen verschiedene Enzyme wie
- Helikasen: entwinden eine überspiralisierte DNS
- Topoisomerasen: heben sowohl die Überspiralisierung als auch die Verdrillung der DNS auf; sie können DNS sowohl zerschneiden als auch wieder zusammenfügen
- Einzelstrangbindeproteine: halten die aufgeschnittene DNS auseinander; so wird ein Ablesen genetischer Informationen erst möglich; diese Proteine stabilisieren also die aufgesplissene DNS
- Polymerasen: sorgen für eine DNS-Kettenverlängerung; sie lassen nach dem Ablesen der Informationen den komplementären DNS-Strang wachsen
- Exonukleasen: entfernen nicht gepaarte oder falsch gepaarte Nukleotide; verläuft die DNS-Kettenverlängerung fehlerhaft (wie beispielsweise durch Falscheinbau einer Base in die DNS oder wenn sich durch eine Basenschädigung keine Wasserstoffbrückenbindungen mehr ausbilden können), so wird die fehlerhafte Base herausgeschnitten und entfernt; Exonukleasen sind also Reparaturenzyme; es gibt auch DNS-Polymerasen mit Exonukleasefähigkeiten
Zytostatika stören vielfach diese Enzyme an unterschiedlichen Stellen, was zu einer fehlerhaften DNS oder deren Genprodukte führt. Vielfach bricht die DNS bei Reparaturversuchen ab (Einzel- oder Doppelstrangbrüche), was das Überleben der Tumorzelle beenden soll.
3.2 Einteilung der antineoplastischen Substanzen
Bei der Einteilung der Substanzen in Klassen sind unterschiedliche Systematiken möglich. Die WHO und daran angelehnt auch die Rote Liste® teilen – historisch nach ihrer Entdeckung bzw. der erstmaligen qualitativen Beschreibung einzelner Wirkausprägungen bedingt – die Substanzen heterogen zum einen nach Wirkungsmechanismus (z. B. Alkylans) und zum anderen nach Ursprung oder Herkunft ein (z. B. Pflanzenalkaloide oder Antitumorantibiotika). Das Klassifikationskriterium „Herkunft“ ist jedoch aus pharmakologischer Sicht wenig hilfreich und würde nur eine riesige Gruppe heterogener Substanzen generieren.
3.2.1 Klassische vs. zielgerichtete Tumortherapeutika
3.2.1.1 Klassische Zytostatika
Die ersten Zytostatika waren Alkylanzien und Antimetabolite. Also chemisch definierte, kleine Moleküle, die Mitte bis Ende der 1940er-Jahre zum ersten Mal am Menschen zum Einsatz kamen. Es gesellten sich Naturstoffe und deren Derivate wie Anthrazykline, Vinkaalkaloide, Podophyllotoxinderivate wie Eto- und Teniposid, sowie Einzelsubstanzen wie Bleomycin und Mitomycin dazu. 1966 wurde der Nobelpreis für den Nachweis vergeben, dass Hormonbehandlungen bei Prostatakrebs machbar und zielführend sind. Diese bahnbrechende Arbeit führte zur Entwicklung von wichtigen neuen Therapieoptionen bei Prostata- und Brustkrebs. Bis zu den 1990er-Jahren wurden dann noch die Platinoide, die Taxane und Topoisomerase-I-Inhibitoren (Irino- & Topotecan) als Substanzklassen in die onkologische Therapie eingeführt. Neben der Einführung einzelner, teilweise Analogsubstanzen, stagnierte die Entwicklung bis etwa zur Jahrtausendwende. Ende der 1990er-Jahre wurden die ersten therapeutischen monoklonalen Antikörper eingeführt Rituximab), zu Beginn der 2000er-Jahre die ersten Kinase-Inhibitoren (Imatinib 2003).
Somit werden, historisch bedingt, folgende Substanzklassen, überwiegend auf Grund ihrer Wirkqualitäten, zu den „klassischen“ Zytostatika gerechnet:
- Alkylanzien
- Antimetabolite
- Mitosehemmer / Spindelgifte
- Topoisomerase-Inhibitoren
- Immunmodulatorisch wirkende Substanzen (IMiD®s)
- Hormone und -Antagonisten sowie verwandte Verbindungen
- Zytotoxische Zytokine
- Interferone
- Interleukine
- Sonstige
- Asparaginasen
- Radikalbildner
- Bleomycin
- Ribonukleodidreduktade-Inhibitoren
- Hydroxyharnstoff = Hydroxycarbamid = Hydroxyurea
- Weitere, nicht näher zuzuordnende, chemisch definierte Substanzen
- Arsentrioxid
- Mifamurtid
- Mitotan
- Tretionin
Auf Subklassifizierungen der einzelnen Klassen wird im Folgenden noch eingegangen, sofern vorhanden.
Die folgenden Säulen zeigen die zeitliche Entwicklung der Zytostatika(-klassen), ohne Anspruch auf Vollständigkeit. Mit der Einführung monoklonaler Antikörper, aber vor allem mit den Kinase-Inhibitoren und sonstigen Enzymhemmstoffen, setzte zum Jahrhundertbeginn eine rasante Entwicklung ein.
| 1946–1960 | 1970 | 1990 | 2008 |
| Stickstoff-Lost Methotrexat 5-Fluorouracil 6-Mercaptopurin | Methotrexat 5-Fluorouracil 6-Mercaptopurin Cyclophosphamid | Methotrexat 5-Fluorouracil 6-Mercaptopurin Cyclophosphamid | Methotrexat 5-Fluorouracil 6-Mercaptopurin Cyclophosphamid |
| Cyclophosphamid | Cytosinarabinosid Daunorubicin Doxorubicin Bleomycin Vincristin Vinblastin Vindesine | Cytosinarabinosid Daunorubicin Doxorubicin Mitoxantron Bleomycin Vincristin Vindesine Etoposid Teniposid Cisplatin Carboplatin 2-Chorodesoxyadenosin Desoxycoformicin Fludarabin Gemcitabin Paclitaxel Docetaxel Vinorelbin Topotecan Irinotecan | Cytosinarabinosid Daunorubicin Doxorubicin Mitoxantron Bleomycin Vincristin Vindesine Etoposid Teniposid Cisplatin Carboplatin 2-Chorodesoxyadenosin Desoxycoformicin Fludarabin Gemcitabin Capecitabin Paclitaxel Docetaxel Vinorelbin Topotecan Irinotecan Pemetrexed Oxaliplatin Rituximab Cetuximab Gefitinib Imatinib Dasatinib Nilotinib Sunitinib Sorafinib Lapatinib Bortezomib Bevacizumab Trastuzumab Alemtuzumab Ibritumumab-Tiuxetan Thalidomid Lenalidomid 5-Azacytidin Decitabin |
Das Grundprinzip der (klassischen) Chemotherapie ist bis heute unverändert: wie die Ursprungssubstanz Senfgas sind all diese Medikamente Zellgifte, die Krebszellen abtöten oder in ihrem Wachstum hemmen sollen. Dabei nutzen die meisten Substanzen die hohe Teilungsrate und genetische Instabilität von Tumorzellen, die diese empfindlicher als gesundes Gewebe reagieren lässt. Allerdings bleiben oft auch normale Zelltypen, die sich ähnlich schnell vermehren – Schleimhaut, Haarwurzelzellen oder blutbildende Zellen – nicht von der schädigenden Wirkung verschont (siehe Abschnitt Zytostatische Chemotherapie). Mit dem Begriff Zellgift war lange Zeit die Vorstellung verbunden, die klassischen Zytostatika hätten kein definierbares molekulares Ziel. Es ist jedoch so, dass jede wirksame Therapie ein molekulares Ziel hat. Im Fall der Zytostatika sind es vielfach Enzyme, die blockiert werden oder andere, funktionelle Zellkomponenten wie DNS oder das Tubilin. Beispiele für Enzymhemmungen durch klassische Tumortherapeutika zeigt die Tabelle unterhalb.
| Gehemmtes Enzym | Substanz |
| Adenosindesaminase (ADA) | Cladribin |
| Fludarabin | |
| Pentostatin | |
| Dihydrofolatreduktase (DHFR) | Methotrexat |
| Pemetrexed | |
| Topoisomerase I | Irinotecan |
| Topotecan | |
| Topoisomerase II | Anthrazykline |
| Podophyllotoxine (Etoposid) |
3.2.1.2 Zielgerichtete Tumortherapeutika (Targeted Therapies)
Mit der Einführung der ersten monoklonalen Antikörper (moAK) wurden meist tumorspezifische Veränderungen (spezielle Tumorantigene oder Überexpression von physiologischen Strukturen, die dem Tumor einen Wachstumsvorteil bieten wie z. B. der EGFR) attackiert. Also Strukturen, die die gesunde, normale Zelle nicht oder nicht in der Menge hat. Diese Definition trifft beispielsweise schon nicht mehr für Antikörper zu, die Wachstumsfaktoren aus dem peripheren Blut abfangen (Aflibercept, Bevacizumab), da diese ja nicht tumorspezifisch sind. Dennoch waren schnell Begriffe wie tumorspezifische Antitumortherapie oder zielgerichtete Therapie – neudeutsch Targeted Therapy – geprägt. Mit der Erfindung von Imatinib hatte man den ersten Hemmstoff eines tumorspezifischen Signaltransduktionswegs. Damit war der Begriff molekular zielgerichtete Therapie aus dem Sprachgebrauch nicht mehr zu eliminieren. Er brachte auch diverse Vorurteile dieser neuen Medikamentenklasse mit sich, die ja im Vergleich zu den monoklonalen Antikörpern ein kleines Molekulargewicht haben und daher auch niedermolekulare Kinase-Inhibtoren bezeichnet werden (nmKI, im Englischen smKI = small molecular Kinase Inhibitor). Die Vorurteile dieser neuen Substanzklasse waren:
- Sie sind hochgradig selektiv – vornehmlich gegen die Tumore
- Sie sind hoch spezifisch – vornehmlich gegen die Tumore
- Sie sind frei von Nebenwirkungen und somit „nicht schädlich“
- Dabei sind sie antitumoral wirksam
- Sie können die konventionelle Chemotherapie ablösen, wir bekommen nach und nach eine „milde“, gut verträgliche und untoxische Antitumortherapie
Die Ernüchterung kam schnell. Während die Fachinformationen der klassischen Zytostatika und anderer Medikamente zu der Zeit durchschnittlich ein dreiseitiges, doppelseitig bedrucktes Faltblatt waren, umfasste die Fachinformation von Imatinib 7 Seiten, von denen viele Seiten Nebenwirkungen und Warnhinweise abhandelten. Wie kommt das? Auch wenn es durch Mutationen tumorspezifische Signalwege gibt, so ist auch die Selektivität der „zielgerichteten“ Therapeutika eine relative. Auf Grund struktureller Ähnlichkeit mit den Normalvarianten der gehemmten Kinasen werden weitere als nur die eigentlich gewünschte, erkrankungsspezifische Kinase gehemmt. Man spricht von sog. „Off-Target-Kinasen“ oder „Bystander-Kinasen“. Insbesondere Multikinase-Inhibitoren hemmen mehr als nur eine Zielkinase. Sie werden teilweise spöttisch aus als Dirty Drug bezeichnet, weil sie eben nicht selektiv wirken. Dies ist auch mit weiteren neuen und neuartigen Toxizitäten verbunden (siehe Teil Nebenwirkungen). Im Zusammenhang mit den Targeted Therapies hört man auch in den Fachkreisen von chemotherapiefreier Tumortherapie. Trotz des neueren Wirkungsmechanismus und der scheinbar größeren Selektivität bleibt unter dem Strich: auch Kinasehemmer machen Zellen tot (sollen sie ja auch) – sind also per definitionem Zytostatika.
Unter den zielgerichteten Therapeutika versteht man also die monoklonalen Antikörper mit großer Molekülmasse und die chemisch definierten, kleinen Moleküle. Da die überwiegende Zahl von ihnen Hemmstoffe von Tyrosinkinasen sind, werden sie häufig pauschal als TKI – Tyrosinkinase-Inhibitoren- bezeichnet. Es gibt jedoch auch Serin-/Threoninkinase-Inhibitoren, daher ist der Begriff nmKI treffender. Weitere Hemmstoffe anderer Enzyme oder Regulatorproteine sind hinzugekommen.
Im Folgenden steht die Bemühung im Vordergrund, Substanzklassen weitestgehend aufgrund des Wirkungsmechanismus der Substanzen (z. B. Alkylierung) oder des Zielmoleküls (z. B. „tubulinaktive“ Agenzien) darzustellen. Es darf dabei nicht unerwähnt bleiben, dass es sich immer um den (grob qualitativen) Hauptwirkungsmechanismus handelt, da einzelne Substanzen mehrere, unterschiedliche Wirkqualitäten bzw. Angriffspunkte aufweisen können, die sich aber erst im Laufe des sich kontinuierlich weiterentwickelnden molekularbiologisch-medizinischen Fortschritts zeigen.
An dieser Stelle sei bereits vorweggenommen, dass auch dieses Konzept (leider) nicht vollständig konsequent durchgehalten werden kann. Bei den antitumoral eingesetzten monoklonalen Antikörpern handelt es sich um eine Klasse von Substanzen, die nicht mit den klassischen, chemisch streng definierten Agenzien vergleichbar ist. Die Gruppe der therapeutisch eingesetzten monoklonalen Antikörper zielt auf unterschiedliche, sogenannte „kritische Zielmoleküle“ aus Sicht des Tumorwirts ab. Von ihnen werden nach und nach die genauen Wirkungsmechanismen bekannt. Diese können beispielsweise von klassischen, immunologischen Mechanismen, wie der antikörperabhängigen zellulären Zytotoxizität (ADCC) und der komplementabhängigen Zytotoxizität (CDC), über einen reinen Rezeptorantagonismus bis hin zur Signaltransduktionsinhibition oder -modifikation reichen. Bei den monoklonalen Antikörpern handelt es sich also um eine Stoffklasse (Proteine). Ebenfalls Proteine sind gegen Tumoren eingesetzte Zytokine (Interleukine, Interferone), sogenannte Biomodulatoren, und Enzyme wie die Asparaginase(n).
3.3 „Klassische“ Zytostatika – Substanzklassen
3.3.1 Alkylanzien
Alkylanzien (alkylierende Verbindungen) sind eine chemisch inhomogene Gruppe von fast ausschließlich organischen Molekülen mit reaktiven Gruppen (Kohlenwasserstoffreste). Die Ausnahme hiervon bilden die Platinderivate, bei denen das zentrale Platinatom das aktive Zentrum darstellt.
Die reaktiven Gruppen der Alkylanzien können intrazellulär leicht mit funktionellen Gruppen reagieren, wie z. B. den Phosphatgruppen der DNS oder Carbonylgruppen von Proteinen. Die bedeutendsten Reaktionen im Hinblick auf eine antitumorale Wirkung sind Alkylierungsreaktionen mit DNS- oder RNS-Basen. Es resultieren (Punkt-)Mutationen (DNS) und Funktionsstörungen (Proteine) durch die chemische Veränderung (monofunktionale Alkylanzien). Durch bi- und höherfunktionale Alkylanzien kann es zu Quervernetzungen von DNS und Funktionsproteinen kommen.
Funktionelle Makromoleküle (z. B. Enzyme) werden inaktiviert. Durch die Zerstörung der DNS-Struktur (Verlust der Matritzenfunktion) wird die Replikationsfähigkeit aufgehoben. Durch die Quervernetzung von DNS-Doppelsträngen werden insbesondere proliferierende Zellen geschädigt. Der Zelltod wird in der Folge durch irreversible DNS-Schädigung (DNS- oder Chromosomenstrangbrüche durch misslingende Reparaturversuche) und/oder gestörte RNS- und Proteinbiosynthese bedingt. Wenn auch die Alkylanzien in allen Phasen des Zellzyklus wirken, so reagieren Zellen insbesondere in der G1-, S- und G2-Phase am empfindlichsten. Innerhalb der Alkylanzien kann aufgrund der chemischen Struktur eine weitere Unterteilung vorgenommen werden. Am Beispiel von Bendamustin wird im Folgenden die bivalente DNS- oder RNS-Basenalkylierung erläutert.
Bendamustin hat als reaktive Partialstruktur eine Bis-chlorethylamingruppe. Durch Chloridabspaltung entsteht im ersten Schritt ein extrem reaktives, instabiles Carboniumion. Dieses reagiert mit beispielsweise einer DNS- und/oder RNS-Base. Nach dieser monovalenten Alkylierung kann sich das Ganze durch Abgang des zweiten Chloridions wiederholen und eine weitere Base alkylieren. So kann die DNS oder RNS innerhalb eines Stranges vernetzt werden (intrastrang Alkylierung). Werden die zwei sich gegenüberliegenden DNS- oder RNS-Stränge vernetzt, spricht man von interstrang Alkylierung oder „Cross Linking“. Sämtliche Platinderivate wirken als reine Cross-Linker.
Etwas atypisch für Alkylanzien verhalten sich die sog. DNA minor groove binding, alkylating agents. Das ursprünglich aus der Ecteinascidia turbinata (eine Seescheidenart) isolierte Trabectedin, gefolgt von Lurbinectedin, binden/alkylieren bevorzugt an Gunaninresten in der kleinen Furche (minor groove) der DNS. Aus dieser Adduktbildung resultiert eine Beugung der DNS-Helix in Richtung großer Furche (major groove). Das führt zu einer Kaskade von Ereignissen, die die transkriptionelle Aktivierung von induzierbaren Genen blockiert und die Formation von Doppelstrangbrüchen induziert. Darauf folgt die Apoptose nach Versagen der DNS-Reparatur durch zelluläre transkriptionsgekoppelte Nukleotidexzisionsreparatur. Trabectedin und Lurbinectedin bilden zudem an ihrem kovalenten Bindungsort eine Vielzahl von Wasserstoffbrückenbindungen zu benachbarten DNS-Strukturen aus. Dadurch werden die beiden DNA-Stränge fest aneinander gebunden und können nicht mehr aufgetrennt und abgelesen werden.
| Alkylanzien | Wirkstoff (INN) |
| „Klassische“, nicht platinhaltige | |
| Aziridine | Thiotepa |
| Bismethanosulfonate | Busulfan Treosulfan |
| Nitrosoharnstoffe | Carmustin (BCNU) Fotemustin Lomustin (CCNU) Streptozozin |
| N-Lost-Abkömmlinge (Bis-chlorethylamine) | Bendamustin Chlorambucil Estramustinphosphat Melphalan Melphalanflufenamid |
| Substituierte N-Lost Derivate Oxazaphosphorine | Cyclophosphamid Ifosfamid Trofosfamid |
| Triazene | Dacarbazin Temozolomid |
| Platinhaltige Verbindungen (reine „Cross-Linker“) | Carboplatin Cisplatin Oxaliplatin |
| DNA Minor Goove Binding Alkylating Agents DMGBAA – Ecteinascidine | Lurbinectidin Trabectidin |
| Sonstige alkylierende Verbindungen | Mitomycin C Procarbazin |
3.3.2 Antimetabolite
Die differenzierte Einteilung der Substanzen aus der Klasse der Antimetaboliten kann einmal aufgrund struktureller Ähnlichkeiten mit physiologisch vorkommenden Substraten (z. B. Purinanaloga) oder nach Wirkqualität (z. B. Enzymhemmstoff) erfolgen. Primär orientiert man sich bei der Benennung von Antimetaboliten üblicherweise an der strukturellen Ähnlichkeit zum physiologischen Substrat (siehe Antifolat, Purin-, Pyrimidinanalogon). Ganz allgemein handelt es sich bei den Antimetaboliten um pharmakologisch und molekular wirksame Verbindungen, die in den Zellstoffwechsel eingreifen. Sie stören Reaktionen mit Schlüsselenzymen von Synthesereaktionen, insbesondere für die DNS- und RNS- bzw. für die daraus resultierende Proteinbiosynthese, indem sie diesen Schlüsselenzymen als physiologisch ähnliches, aber letztendlich nicht funktionelles oder „falsches“ Substrat oder Intermediat zur Reaktion angeboten werden.
Die in der antitumoralen Therapie eingesetzten Antimetaboliten beeinflussen im Wesentlichen die Nukleinsäuresynthese (DNS, RNS). Durch die Hemmung bestimmter Enzyme, z. B. der Adenosindesaminase, können bestimmte Antimetaboliten zu zytotoxischen (lymphotoxischen) Metaboliten kumulieren.
Zuletzt in der Klasse der (formalchemisch) Antimetaboliten hinzugekommen sind mit Stickstoff substituierte (Pseudo-)Pyrimidinanaloga, sogenannte Triazine. Ein über die oben genannte pharmakodynamische Wirkqualität hinausgehender (Haupt-)Wirkungsmechanismus dieser beiden Substanzen ist eine Hemmung der DNS-Methyltransferase (siehe DNMTi). Resultat ist eine DNS-Hypomethylierung. Diese Hypomethylierung soll zur Wiederherstellung der krebsunterdrückenden Funktionen von Genen bei Krebszellen führen. Die Hypomethylierung kann Genfunktionen restaurieren, die zur Differenzierung zu normalen Zellen notwendig sind. Mit anderen Worten: Es sollen hypermethylierte und damit stillgelegte Tumorsuppressorgene wieder reaktiviert werden. Man möchte also die Schutzfunktion wieder herstellen. Wie bei den HDACi (Hemmstoffe der Histondesacetylase) handelt es sich um einen epigenetischen Wirkansatz[2]. Wirkstoffe dieser Klasse sind Azacitidin und Decitabin. Ob diese Pharmakodynamik auf Triazine beschränkt ist, ist derzeit noch unklar.
| Antimetabolite | Wirkstoff (INN) |
| „Antifolate“ | Methotrexat = Folsäureantagonist Pemetrexed (formals MTA = Multi Target(ed) Antifolate) Raltitrexed (direkter Thymidilatsynthetase-Hemmer) |
| Purinanaloga | 6-Mercaptopurin (6-MP) 6-Thioguanin (6-TG) Cladribin (2-CdA) Clofarabin Fludarabinphosphat Nelarabin Pentostatin; Intermediat (2’-Desoxycoformicin) |
| Pyrimidinanaloga | Capecitabin Cytosinarabinosid 5-Fluorouracil Gemcitabin Tegafur: Gimeracil: Oteracil (vormals S 1) Tegafur: Uracil Trifluridin: Tipiracil |
| (Pseudo-)Pyrimidine Triazine | Azacitidin Decitabin |
3.3.3 Inhibitoren des mikrotubulären Systems (Mitosehemmer/Spindelgifte)
Mikrotubuli sind integraler Bestandteil der Kernspindel, die sich während der Zellkernteilung bildet. Die Mikrotubuli sind röhrenförmige intrazelluläre Proteinstrukturen zur Zellstabilisierung des Zytoskeletts. Durch die Verkürzung der Tubuli während der Anaphase kommt es zur Verlagerung der Chromosomen zu den Zentren der beiden entstehenden Zellen im Rahmen der Zellteilung. In diese Gruppe von tubulinaktiven Agenzien fallen die Vinca-Alkaloide, die Taxane, die Halichondrine und die Epothilone (Zulassung derzeit nur in der Schweiz und den USA).
Vinca-Alkaloide: Das Wirkprinzip der tubulindestruierenden Vinca-Alkaloide ist eine Interaktion mit Tubulin in Form einer Unterbrechung der Tubulinpolymerisation und einer Zerstörung bereits polymerisierten Tubulins. Diese Zerstörung des Spindelapparates arretiert die Zellteilung in der Metaphase.
Taxane: Die derzeit zur Verfügung stehenden tubulinstabilisierenden Taxane binden scheinbar an die gleichen Stellen der Tubulinpolymere. Sie stabilisieren die Mikrotubuli. Der Abbau eines bestehenden Zytoskeletts wird verhindert, wodurch der Mitosevorgang arretiert bzw. unterbrochen wird. Durch die sich nur noch verlängernden Mikrotubuli entstehen in der Zelle sternförmige Riesenmikrotubuli, was als „eingefrorene Mitose“ (Freezed Mitosis) bezeichnet wird.
Qualitativ gleich, jedoch mit einer anderen Bindungsstelle am beta-Tubulin, wirken die Epothilone (erster zugelassener Wirkstoff ist das Ixabepilon, auch Aza-Epothilon B). Wegen der nicht überlappenden Bindung und Wirkung auf Tubulin-Isoformen sollen die Epothilone auch bei Taxanresistenz wirksam sein. Zudem sind sie ein deutlich schlechteres Substrat für P-Glykoprotein 170.
Halichondrine: Eine Art Zwischenstellung zwischen Vinca-Alkaloiden und Taxanen nehmen die Halichondrine ein. Eribulin behindert am wachsenden Ende die Tubulinpolymerisation, verhindert aber nicht den Abbau/Umbau am anderen Ende. Das Mikrotubulin wird in Bruchstücke (Segmente) zerlegt.
| Inhibitoren des mikrotubulinären Systems auch Mitosehemmer/Spindelgifte | Wirkstoff (INN) |
| Tubulinstabilisierende Agenzien – Taxane | Cabazitaxel Docetaxel Paclitaxel |
| – Epothilone | Ixabepilon |
| – Halichondrine | Eribulin |
| Tubulindestruierende Agenzien Vincaalkaloide | Vinblastin Vincristin Vindesin Vinflunin Vinorelbin |
3.3.4 Topoisomerase(TOP)-Hemmstoffe
Topoisomerasen (TOP) sind Enzyme, die die räumliche Struktur der DNS kontrollieren und modifizieren. Das beinhaltet, dass diese Enzyme die DNS spalten können, und umfasst vorübergehende DNS-Einzel- und DNS-Doppelstrangbrüche. Damit sind Topoisomerasen zunächst in der Lage, sowohl die Überspiralisierung als auch die Verdrillung der DNS aufzuheben. Sie beseitigen dadurch die topologischen Sperren der hochgradig im Zellkern gefalteten DNS, um ein Ablesen einzelner Gene zu ermöglichen oder die Voraussetzungen zur DNS-Duplikation zu schaffen. TOP können aufgetrennte DNS-Fragmente auch wieder verbinden. Sie sind somit verantwortlich für das sogenannte „supercoiling“ und „twisting“, also das Verdrillen, Verknoten und Überspiralisieren der DNS, damit diese nach dem Ablesen der benötigten Informationen wieder komprimiert im Zellkern vorliegt. Es sind nukleäre Enzyme mit essenzieller Bedeutung für Replikation, Transkription und DNS-Reparatur. In Säugerzellen können zwei Typen von Topoisomerasen differenziert werden, die TOP I und die TOP II. Topoisomerase I induziert Einzelstrangbrüche für die sogenannte Replikationsgabel. Die DNS wird so entspiralisiert und ist für die informationslesenden und -verarbeitenden Enzyme sterisch zugänglich. Nach Ablesen der benötigten Informationen wird die Phosphodiesterbindung wieder hergestellt und die Topoisomerase dissoziiert wieder. Zur Entdrillung und zur Verkürzung der DNS wird die Topoisomerase II benötigt.
TOP-I-Hemmstoffe
Die TOP-I-Hemmstoffe (Tecane) binden an den Spaltungskomplex, stabilisieren diesen und verhindern einerseits die Religation, was zu DNS-Einzelstrangbrüchen führt. Andererseits kann die TOP I nicht mehr von der DNA freigesetzt werden und es resultiert eine Interaktion mit dem fortschreitenden DNS-Replikationskomplex. Die Zelle kann nicht anders, als auch den zweiten DNS-Strang zu durchtrennen. Die Folge dieser Interaktion ist ein Doppelstrangbruch.
TOP-II-Hemmstoffe
Hemmstoffe der Topoisomerase II sind das Podophyllotoxinderivat Etoposid sowie, indirekt, Anthrazykline und verwandte Verbindungen. Um die oben erwähnte DNS-Entdrillung und eine Strangpassage im Sinne eines Entknotens der chromosomalen DNS zu ermöglichen, wird die doppelsträngige DNS vorübergehend von der dimeren TOP II gespalten. Die Podophyllotoxinderivate binden an die TOP II (nicht an die DNS) und bewirken einerseits eine Hemmung der katalytischen Aktivität und andererseits eine Stabilisierung dieses Doppelstrang-Spaltungskomplexes. Durch Proteinspaltung und Denaturierung kommt es im Weiteren zu persistierenden Doppelstrangbrüchen und Zelltod. Aufgrund dieses Mechanismus werden die Podophyllotoxinderivate auch nicht-interkalierende Topoisomerase-II-Hemmer genannt. Podophyllotoxinderivate sind leukämogen.
Analog dazu gibt es auch interkalierende Topoisomerase-II-Hemmer. Sie besitzen ober- und unterhalb ihres aromatischen Ringsystems leicht polarisierbare Elektronenwolken und sind dadurch in der Lage, sich zwischen übereinander gestapelte Basenpaare der Nukleinsäuren zu schieben. Durch starke intermolekulare Kräfte können die DNS-Stränge nicht mehr zur Replikation voneinander getrennt werden (elektrostatische Quervernetzung), so dass es dem Replikationsgabelkomplex nicht möglich ist, die entsprechenden DNS-Abschnitte abzulesen. So ist ein postulierter Mechanismus, dass durch Interkalation die dreidimensionale DNS-Konfiguration verändert wird und ein Zusammenfügen des temporären Doppelstrangbruchs für die TOP II unmöglich wird. Daher spricht man auch von indirekter TOP II-Hemmung. Als eine Art Nettoeffekt verschieben sie quasi die Hin- und Rückreaktion der Topoisomerasen in Richtung irreversibler DNS-Fragmentierung. Diese Substanzen werden auch als Interkalanzien oder Matrizenblocker bezeichnet. Letzteres, da die DNS als „Lesevorlage“ – als Matritze – nicht mehr abgelesen werden kann.
| Topoisomerase-Hemmstoffe | Wirkstoff (INN) |
| Hemmstoffe der Topoisomerase I | Irinotecan Topotecan |
| Hemmstoffe der Topoisomerase II | |
| nicht interkalierende | Etoposid |
| interkalierende indirekt hemmende; Matritzenblocker | Amsacrin Dactinomycin Daunorubicin Doxorubicin Epirubicin Idarubicin Mitoxantron Pixantron |
3.3.5 Immunmodulatorisch wirkende Substanzen (IMiDs®)
IMiD® steht für Immune Modulatory imide Drug, und ist einmal von der chemischen Partialstruktur -Imid- und ihrer Wirkung abgeleitet. IMiD® ist ein geschützter Ausdruck.
Die derzeitigen IMiDs® sind oral anwendbare niedermolekulare Wirkstoffe, deren chemische Struktur auf Thalidomid basiert. Neben Thalidomid werden derzeit Lenalidomid und Pomalidomid eingesetzt. Es handelt sich um multifunktionale Wirkstoffe, die die Produktion diverser Zytokine im Mikromilieu von Tumoren beeinflussen. So wird beispielsweise die Synthese der proinflammatorischen Zytokine TNF-α, IL-1β, IL-6 und IL-12 gehemmt, die des antiinflammatorischen IL-10 dagegen gesteigert. Qualitativ sind folgende weitere Wirkmechansimen feststellbar:
- Hemmung der Proliferation bestimmter hämatopoetischer Tumorzellen
- Stimulation von T-Zellen und natürlichen Killerzellen (NK-Zellen) und in Folge Verbesserung der durch diese Zellen vermittelten Immunität gegen Tumorzellen
- Erhöhung der natürlichen Killer-T-Zellen
- Steigerung der Blutbildung (Erythropoese)
- Antiangiogene Eigenschaften (siehe auch Abschnitt Wirkprinzip Antiangiogenese)
Diese qualitativen Beschreibungen sagen jedoch nichts darüber aus, auf Grund welcher molekularen Ziele dies geschieht. Auf molekularer Ebene wirken die IMiDs® dadurch, dass sie zur selektiven Ubiquitinierung zweier lymphoider Transkriptionsfaktoren (IKZF1 und IKZF3) durch die CRBN-CRL4 Ubiquitinligase führen. Das Anfügen von Ubiquitin – ein kleines Protein – an Signalproteine ist oft, aber nicht nur, ein zelluläres Signal mit der Botschaft: „Kann abgebaut werden / wird nicht mehr benötigt“. Der Abbau geschieht im Proteasom (siehe Proteasom-Inhibitoren). Vom Lenalidomid weiß man, dass es die Proteinkinase Caseinkinase-1-Alpha (CK1α) zur Ubiquitinierung bringt. Sie wird ebenfalls im Proteasom abgebaut. Die CK1α ist eine Serin-/Threoninknase, die TP53 aktiviert, das wiederum antiapoptotisch wirkt. Darüber aktiviert sie den onkogenen Wnt/β-Catenin-Signalweg. Das Gen für CK1α heißt CSNK1A1 und befindet sich auf Chromosom 5 im Abschnitt 5q. Dies erklärt, warum Lenalidomid bei einem myelodysplastischen Syndrom mit 5q-Deletion (del[5q]) und entsprechender Haploinsuffizienz besonders gut wirkt.
All diese Wirkmechanismen gelten als Grundlage für eine neue Form von therapeutischen Wirkstoffen, den sogenannten PROTACs (Proteolysis Targeting Chimeras). Diese führen zum selektiven Abbau von Zielproteinen – üblicherweise mit Signalfunktion – durch Ubiquitinierung.
Der Arzneistoff Thalidomid und seine Schwesterverbindungen binden auch an Cereblon. Cereblon ist ein Protein, das im Menschen in vielen Gewebetypen exprimiert wird, so z. B. im Gehirn. Dort stabilisiert es in der Zellmembran bestimmte Kaliumkanäle und trägt so zum Lernen und zur Funktion des Gedächtnisses bei. Mutationen im CRBN-Gen können zu erblicher milder Demenz aufgrund von Lern- und Gedächtnisstörungen führen. Cereblon besitzt weitere, wichtige Schlüsselfunktionen während der Embryonalentwicklung. Es bildet an einer substratbindenden Untereinheit einer Ubiquitinligase, die über die Regulation der Transkription das Wachsen von Gliedmaßen beeinflusst. Die IMiDs® binden (auch) an Cerebolon und stören wahrscheinlich so das normale Wachstum des Embryos (→ Contergan®-Skandal). Die Verschreibung der IMiDs® erfolgt wegen ihres starken Missbildungspotenzials dokumentationspflichtig und streng überwacht mittels sog. T-Rezepte.
3.3.6 Hormone, Hormonantagonisten und verwandte Verbindungen
Substanzen dieses Typs werden bei sogenannten hormon- bzw. endokrin sensitiven Tumoren, wie z. B. Mamma-, Prostata-, Korpus-, Nieren-, Pankreas- oder Endometriumkarzinomen eingesetzt. Eine weitere Tumorentität für diese Wirkstoffe ist das Karzinoidsyndrom. Das Wachstum lässt sich durch Zufuhr oder Entzug von Hormonen bremsen. Die eingesetzten Substanzen können hormonagonistische oder -antagonistische Wirkungen entfalten. Grundsätzlich können körpereigene wachstumsfördernde Hormone durch Antagonisten von ihrem Wirkort verdrängt werden (kompetitive Wirkung z. B. Antiöstrogene, -gestagene, -androgene). Ein anderer Angriffspunkt ist die Hemmung der Hormonbiosynthese, z. B. durch Aromatasehemmer, sodass keine Agonisten synthetisiert werden können (inhibierende hormonelle Wirkung). Die Aromatasehemmstoffe unterbinden den letzten Schritt bei der Biosynthese von Östrogenen (Östradiol, Östron), ohne mit anderen Schritten der Steroidhormonbiosynthese zu interferieren. Weiterhin nutzt man durch Anwendung hochdosierter, gleichgeschlechtlicher Hormone eine Hormoninterferenz, indem negative Rückkoppelungsmechanismen ausgelöst werden.
| Hormone, Hormonantagonisten und verwandte Verbindungen | Wirkstoff (INN) |
| Antiandrogene | Abirateron Apalutamid Bicalutamid Cyproteronacetat Darolutamid Enzalutamid Flutamid |
| Antiöstrogene | Tamoxifen |
| Aromatasehemmer | Anastrozol Exemestan Letrozol |
| Gestagene | Keine mehr im Handel |
| Östrogene | Keine mehr im Handel |
| Superagonisten (LHRH Agonisten, auch GnRH Analoga) | Buserelin Goserelin Leuprorelin Triptorelin |
| GnRH Rezeptorantagonisten | Relugolix |
3.3.7 Zytotoxische Zytokine
Zytokine ist der Oberbegriff für zahlreiche, von einer Vielzahl von Zellarten gebildeten und sezernierten Substanzen, die als interzelluläre Mediatoren zur Aktivierung von Zellen beitragen. Andere Umschreibungen und Begriffe sind: Lymphokine, Interleukine, Monokine und Wachstumsfaktoren oder – wie eingangs erwähnt – Biomodulatoren. Zytokine sind körpereigene Eiweißsubstanzen (Peptide), die beispielsweise auch von aktivierten T-Zellen und anderen Zellen während der natürlichen und der spezifischen Immunantwort freigesetzt werden. Sie haben vielfältige steuernde Funktionen, wie Proinflammation, Immunoregulation und auch Steuerung der Hämatopoese. Zu den therapeutisch eingesetzten, zytotoxischen und antitumoralen Zytokinen gehören die Interleukine (einzige derzeit klinisch eingesetzte Substanz ist das IL-2) und Interferone.
| Zytotoxische Zytokine | Wirkstoff (INN) |
| Interferone | Interferon-alpha-2a Interferon-alpha-2b Interferon-beta |
| Interleukine | Aldesleukin (Interleukin-2) |
3.3.8 Sonstige
Enzyme (Asparaginase, Crisantaspase = Erwinase, PEG-Asparaginase): Bestimmte Tumorzellen, insbesondere leukämische Lymphoblasten, sind von der Aminosäure Asparagin abhängig, da ihnen die Asparagin-Synthetase fehlt. Sie sind nicht in der Lage, Asparagin aus Glycin zu bilden. Das Enzym Asparaginase (L-Asparagin-Amidohydrolase) ist ein zytostatisch wirksames Enzym, das spezifisch L-Asparagin in L-Asparaginsäure und Ammoniak spaltet. Durch den Einsatz von Asparaginase verarmen die Tumorzellen an der für sie essenziellen Aminosäure. Die Folge ist eine Hemmung der Proteinbiosynthese.
DNS-spaltende Agenzien (Radikalbildner): Bleomycin z. B. mediiert auch die Entstehung von Radikalen. Diese gebildeten Radikale führen über eine Art Kettenreaktion letztendlich zu einer Spaltung der DNS-Einzel- und DNS-Doppelstränge. Durch Interkalation inhibiert Bleomycin in geringem Ausmaß auch die DNS-Polymerase und behindert die DNS-Reparatur.
Ribonukleotidreduktase-Inhibitoren: Die einzige therapeutisch eingesetzte Substanz, die dieses Enzym hemmt, ist das Hydroxycarbamid. Durch eine spezifische Hemmung der Nukleosiddiphosphatreduktase wird die DNS-Synthese gestört. Die Bildung von Desoxynukleosidtriphosphaten wird unterbunden, eine DNS-Reparatur verhindert.
3.3.9 Weitere, nicht näher zuzuordnende, chemisch definierte Substanzen
Arsentrioxid: Arsenverbindungen induzieren Chromosomenschäden und morphologische Zellveränderungen und gelten beim Menschen als kanzerogen. Der therapeutische Einsatz ist die refraktäre oder auf Standardtherapeutika nicht ansprechende akute Promyelozytenleukämie. Der genaue Wirkmechanismus von Arsentrioxid ist noch nicht geklärt, es existieren jedoch zwei Hypothesen:
- Schädigung des abnormalen Proteins (ein Fusionsprotein), welches die Myelozytenreifung stört
- proapoptotische Wirkung, indem die Freisetzung von Caspase-Enzymen gefördert wird
Mifamurtid: Auch Muramyltripeptid-Phosphatidyl-Ethanolamin genannt, ist ein vollsynthetisches Analogon eines Zellwandbestandteils von Mycobacterium sp., dem kleinsten natürlich vorkommenden immunstimulierenden Bestandteil der Zellwand von Mykobakterien. Über komplexe Mechanismen werden Immunzellen (Monozyten/Makrophagen) aktiviert. Hinweis: Mifamurtid wirkt nur in Kombination mit konventioneller Chemotherapie. Es wird im Anschluss an eine makroskopisch vollständige Tumorresektion (hochmaligne Osteosarkome) im Rahmen einer postoperativen Kombinationschemotherapie eingesetzt.
Mitotan: Diese Substanz ähnelt in ihrer chemischen Struktur dem Insektizid DDT (p,p’-Dichlordiphenyltrichloräthen). Es beeinflusst den Metabolismus von Steroiden in der Peripherie und führt zu einer Suppression der Nebennierenrinde. Unter Mitotantherapie kommt es daher zu einer Nebennierenrindenatrophie, begleitet von einem Absinken der Gluko- und Mineralokortikoidsynthese.
Tretionin: Dieser physiologisch vorkommende Vitamin-A-Metabolit induziert in pharmakologischen Dosen die Zelldifferenzierung und hemmt die Zellproliferation von hämatopoetischen Zellen.
3.4 Neuere Substanzklassen (Zulassung um die Jahrtausendwende)
3.4.1 Hemmstoffe von Proteasomen
Das Proteasom ist ein Proteinkomplex aus über 30 verschiedenen Proteinen, der im Zytoplasma und im Zellkern vorkommt. Der Komplex fungiert als Protease (Hydrolase) und fragmentiert Proteine, es ist sozusagen der „Schredder“ der Zelle. Abzubauende (Funktions-)Proteine werden durch Ubiquitinmoleküle gekennzeichnet.
Merke: Das Proteasom ist der „Schredder“ der Zelle.
Durch eine Hemmung des Proteasoms kommt es zu einer Abbauhemmung von regulatorischen Proteinen. Das Boronsäurederivat (kovalente Bindung eines Boratoms mit einem Kohlenstoffatom) mit Proteinpartialstruktur Bortezomib hemmt den Multienzymkomplex reversibel durch Bindung an die Chymotrypsin-ähnliche Untereinheit. Die Folge dieser Hemmung ist eine Akkumulation und Stabilisierung von pro- und antiapoptotischen Proteinen. Durch diese unkoordinierten, widersprüchlichen Informationen kommt es zum Zellzyklusarrest mit nachfolgender Apoptose. Bildlich gesehen „erstickt“ die Zelle an ihren eigenen Proteinen. Maligne Zellen reagieren auf die Proteasom-Hemmung empfindlicher als normale Zellen. Bortezomib beispielsweise hemmt bei den eingesetzten Dosierungen das Proteasom zu ca. maximal 80 Prozent. Eine basale Proteasomaktivität bleibt erhalten, sodass normale Zellen in ihrer Funktionsfähigkeit weniger stark beeinträchtigt werden.
Weitere Wirkmechanismen umfassen die Inhibition der Genexpression mit einer Hemmung des Transkriptionsfaktors NFκB. Bortezomib wirkt durch eine Inhibition der Expression von zellulären Adhäsionsmolekülen antimetastatisch. Funktions- und Signalproteine, die nicht mehr benötigt werden, werden zunächst mit Ubiquitinketten (Ub) gekennzeichnet. Dies ist das Signal für einen proteasomalen Abbau. Wird dieser Abbau durch Proteasom-Inhibitoren blockiert, kommt es in der Folge zu einer Reihe widersprüchlicher Signale für die Zelle. Außerdem kumulieren diese Proteine. Die Zelle erstickt an ihrem eigenen Eiweiß.
Ebenfalls ein Boronsäurederivat ist das nur peroral verfügbare Ixazomib. Carfilzomib verfügt über eine Epoxyketonstruktur und hemmt das Proteasom durch Alkylierung irreversibel. Derzeit unterscheidet man reversible Proteasom-Inhibitoren mit „Boronic Acid“- und irreversible Proteasom-Inhibitoren „Epoxyketone-Warhead“. Die irreversiblen Proteasom-Inhibitoren wirken eigentlich als proteasomspezifische Alkylanzien.
3.4.2 Hemmstoffe der Histondesacetylase (HDACi)
Histone (basische Proteine) dienen primär der Verpackung der DNS im Zellkern. Die DNS ist um die Histone gewickelt, ähnlich einer Schnur um einen Tennisball. Das so durch Histone „komprimierte“ DNS-Molekül ist etwa 40.000-fach kompakter als die unverpackte DNS. Die Histone mit der darum gewundenen DNS werden als Nukleosom bezeichnet. Sie haben eine wichtige Funktion bei der Regulation der Genexpression und der DNS-Reparatur. Solange die DNS um das Histon gewickelt ist, können Genabschnitte nicht abgelesen werden. Sie sind stillgelegt.
Sind durch Deacetylierung Genabschnitte von beispielsweise Tumorsupressorgenen blockiert, verlieren sie ihre Schutzwirkung und Tumore können entstehen und/oder wachsen.
Acetylierung (Übertragung von Essigsäureresten) findet ausschließlich an Lysinresten statt. Damit wird die der positiven Ladung des Lysins neutralisiert. Dies führt zur Herabsetzung der elektrostatischen Wechselwirkungen zwischen Lysin und dem negativ geladenen Rückgrat der DNS. Die DNS wird sozusagen für das Ablesen von Genen aufgeweitet. Die Acetylierung wird durch Histonacetyltransferasen ausgeführt, die Entfernung der Acetylreste von Histondeacetylasen (HDAC). Diese HDAC entfernen im Zellkern die Essigsäurereste (Acetylgruppen) von acetyliertem Lysin auf dem N-terminalen Histonende. Dadurch bekommt Lysin wieder eine positive elektrische Ladung, was die Affinität des Histonendes für das negativ geladene Phosphat-Gerüst der DNS erhöht. Durch die daraus folgende Blockierung der DNS für Transkriptionsfaktoren wird die DNS-Transkription herunterreguliert. Diese Genabschitte werden sozusagen wieder stillgelegt. Daraus resultiert eine verminderte oder fehlende Synthese von tumorprotektiven Faktoren, wie z. B. dem Genprodukt des Tumorsuppressorgens p53 (Gen-Silencing). Die erste zugelassene, so wirkende Substanz ist das Vorinostat (Suberoylanilidhydroxamsäure) mit der Indikation kutanes T-Zellen-Lymphom. HDAC-Inhibitoren (HDACi) werden auch als epigenetische Hemmstoffe bezeichnet.
Merke:
Acetylierung → bewirkt Aktivierung von Transkription, Aktivierung ruhig gestellter Gene mit Schutzfunktion
Desacetylierung → Downregulation von Genaktivität, Stilllegung
3.4.3 DNS-Methyltransferase-Inhibitoren – DNMTi
Ähnlich wie eine Desacetylierung können DNS-Abschnitte durch eine Hypermethylierung stumm geschaltet werden. Ein häufiger onkogener Mechanismus ist dabei die Hypermethylierung von Tumorsuppressorgenen. Diese Aktivitätsveränderungen können stabil an Tochterzellen vererbt werden. Das versucht man mit medikamentöser Hemmung der DNS-Methyltransferasen (DNA Methyl Transferases – DNMTs) wieder rückgängig zu machen. Mit diesen DNMT-Inhibitoren (DNMTi) sollen epigenetisch stillgelegte Gene durch Hypomethylierung reaktiviert werden. Daher werden DNMTi auch als hypomethylierende Substanzen genannt. Die ersten beiden zugelassenen Substanzen, Azacitidin und Decitabin, ähneln formalchemisch Antimetaboliten (siehe Abschnitt Antimetabolite). Sie werden teilweise auch in RNS und DNS eingebaut. Der Beitrag zur Zytotoxizität als Antimetabolit ist jedoch unklar.
3.5 Monoklonale Antikörper
3.5.1 Allgemeines zu therapeutisch eingesetzten monoklonalen Antikörpern
Physiologischerweise sind Antikörper eine zu den Gammaglobulinen gehörende heterogene Gruppe von Glykoproteinen (Immunglobuline). Sie stellen eine mögliche Antwort des Immunsystems nach Kontakt des Organismus mit Antigenen dar, werden von B-Lymphozyten und Plasmazellen gebildet und in die Körperflüssigkeiten sezerniert. Als Träger der humoralen Immunität binden Antikörper vornehmlich an fremde (pathogene) Mikroorganismen, aber auch körpereigene Antigene (z. B. Tumorzellen), und können diese neutralisieren. Monoklonale Antikörper (moAK) sind Antikörper, die aus einem Zellklon gebildet werden und daher monospezifisch sind.
Die Rationale einer antitumoralen Therapie mit monoklonalen Antikörpern ist, dass Gewebe von Tumorzellen idealerweise ein oder mehrere spezifische Tumorantigene auf ihrer Oberfläche exprimieren. Dadurch unterscheiden sie sich deutlich von benignem Gewebe. Tumorzellen könnten dann vom Immunsystem als „fremd“ erkannt und zerstört werden. Ein Tumor kann aber auch eine erhöhte Anzahl nicht mutierter, physiologischer Strukturen aufweisen, die ihm dann einen Wachstums- und Überlebensvorteil bieten. So beispielsweise eine erhöhte Anzahl des EGF-Rezeptors auf bestimmten Tumorzellen.
Speziell Anti-EGFR-Strategien: Zu den zellmembranständigen Rezeptoren aus der Familie der epidermalen Wachstumsfaktorrezeptoren gehören die vier verwandten, funktionell unterschiedlichen Homologe EGFR1 bis EGFR4 (synonym: HER1 bis HER4 oder auch ErbB1 bis ErbB4). Die inaktiven Monomere homo- oder heteromerisieren nach extrazellulärer Ligandenbindung. Durch Konformationsänderung kommt es zur Annäherung der intrazellulär gelegenen Tyrosinkinase-Domänen, gefolgt von einer Autophosphorylierung. Durch diese Phosphorylierung werden nachfolgende Signalkaskaden aktiviert, wodurch es zur Zellproliferation und Apoptosehemmung kommt. Erkrankungen mit einer malignitätsassoziierten Überexpression des EGFR1 (HER1) sind beispielsweise das kolorektale Karzinom, das nicht-kleinzellige Bronchialkarzinom und das Pankreaskarzinom. HER2 wird dagegen bei bestimmten Mammakarzinomen überexprimiert. Die Überexpression der Rezeptoren muss nachgewiesen werden (Tumorbiopsie), sonst entbehrt eine Therapie mit Medikamenten gegen diese Zielstrukturen jeglicher Grundlage.
Während Anti-EGFR-Antikörper die Rezeptoren von der extrazellulären Seite blockieren, unterbinden die nmKI die Signaltransduktion an einem intrazellulären Angriffspunkt.
3.5.2 Effektormechanismen
Um die Effektormechanismen zu verstehen, sei im Folgenden in aller Kürze auf die Struktur der moAK eingegangen.
Die zum therapeutischen Einsatz kommenden moAK sind üblicherweise Immunglobuline vom IgG-Typus. Hierbei handelt es sich um das bekannte Modell eines Y-geformten Moleküls, bestehend aus 4 Proteinketten mit zwei leichten, den sog. L-Ketten (L = light) und 2 schweren, sog. H-Ketten (H = heavy), die über Disulfidbrücken miteinander verbunden sind. Das Carbonsäureende der schweren Ketten heißt auch konstante Region oder Fc-Teil (Fc = Fragment Crystalline), während das Aminoende hochvariable Sequenzen enthält, die die Antigen-Erkennungsstelle bilden. Diese Bereiche werden auch als Fab-Region (Fab = Fragment antigen binding) bezeichnet.
Die potenziellen Angriffspunkte und Effektormechanismen einer solchen Antikörpertherapie sind unterschiedlicher Natur. Neben klassisch immunologischen Mechanismen, wie die antikörperabhängige zelluläre Toxizität (ADCC) und die komplementabhängige Zytotoxizität (CDC), spielt auch die Hemmung und/oder Beeinflussung der intrazellulären Signaltransduktion eine Rolle. Monoklonale Antikörper können aber nicht nur Tumorantigene und Rezeptoren blockieren, sondern im Blut lösliche Wachstumsfaktoren abfangen, wie es im Rahmen der antiangiogenen Therapie erfolgt (siehe Abschnitt Wirkprinzip Antiangiogenese). Zur Benennung der Wirkstoffe siehe Abschnitt Namensgebung.
3.5.2.1 ADCC
Bei der ADCC (= Antibody-dependent cellmediated cytotoxicity), zu deutsch antikörperabhängige zellmediierte Zytotoxizität, markiert der moAK sozusagen eine Tumorzelle, indem er an tumorspezifische Oberflächenantigene andockt. Der Antikörper wird von zu natürlichen Killerzellen (NK-Zellen) ausgereiften Lymphozyten erkannt. Durch einen spezifischen Kontakt (NK-Zelle ↔ AK ↔ Zielzelle) dieser als Effektorzellen fungierenden NK-Zellen kommt es durch Freisetzung von lysosomalen Enzymen (Perforine und Granzyme) schlussendlich zur Zelllyse. Neben NK-Zellen vermitteln Monozyten/Makrophagen ebenfalls die ADCC. Antikörper, die mit ihrem Fab-Teil an ein (Zell-)Antigen binden und mit ihrem Fc-Teil Effektorzellen wie NK-Zellen und/oder Monozyten/Makrophagen binden und aktivieren können, werden auch bisspezifische Antikörper genannt.
Bisspezifische Antikörper binden mit ihrem Fab-Teil an das Tumorantigen und meist an den CD3-Rezeptor von T-Zellen. Mit dem Fc-Rezeptor binden sie an NK-Zellen, Makrophagen und/oder dendritische Zellen. Durch Interaktion der Effektorzellen mit der Tumorzelle werden diese Antikörper abgetötet. Der erste therapeutische moAK dieser Klasse war das Blinatumumab. Nicht an den CD3-Rezeptor von T-Zellen bindet Amivantamab. Hier handelt es sich um einen anti-EGFR x anti-MET-Antikörper.
3.5.2.2 CDC
Das Komplement ist ein im Blut gelöstes Proteasesystem und Teil des unspezifischen, humoralen (zellfreien, auf Körpersäfte beruhenden) Immunsystems. Es dient primär zur Eliminierung von zellulären Antigenen (z. B. Bakterien). Die Aktivierung der komplementabhängigen Zytotoxizität (CDC = complement-dependent cytotoxicity) erfolgt durch bestimmte Immunglobulinklassen. Die Komplementbindung erfolgt am sog. Fc-Teil des Antikörpers. Der sog. klassische Komplementpfad wird durch antikörperbedeckte Zielregionen oder durch Antigen-Antikörperkomplexe aktiviert. Der Immunkomplex, bestehend aus an die Tumorzelle gebundener Antikörper, wird vom Komplement erkannt, wodurch die sog. Komplementkaskade abläuft (C1 → C4C2 → C3 → C3bC5 → C5bC6C7C8C9 → C5b-9 = MAC). Durch den am Ende der Kaskade stehenden Membrane Attack Complex (MAC) kommt es zur Porenbildung und somit zur Zelllyse. Eine ältere Bezeichnung für solche Antikörper ist zytotoxische AK.
3.5.3 Weitere Effektormechanismen
Mit Einführung der Anti-CD38-Antikörper wurde die ADCP (= Antibody-dependent cell-mediated phagocytosis), also die Antikörper-induzierte Anlockung von Phagozyten beschrieben. Diese Fresszellen sind zumeist gewebsständige, oft jedoch amöboid bewegliche, zur Phagozytose befähigte Zellen. Sie haben die Fähigkeit, Partikel, Mikroorganismen und Flüssigkeiten aufzunehmen und im Zellinnern zu verdauen, ein bis dahin unbekannter Mechanismus von moAK.
MoAK modulieren auch die intrazelluläre Signalgebung, die dann in der Konsequenz zum Zelltod führt. Diese Signaltransduktionsmodulation wurde und wird z. T. immer noch mit „Induktion der Apoptose“ als eher nebulös wirkende Pharmakodynamik beschrieben.
3.6 Wirkprinzipien von moAK
Die derzeitig verfügbaren Antikörper/Fusionsproteine können wie folgt in 3 Wirkprinzipien eingeteilt werden:
1. Antikörper bindet an eine zelluläre Zielstruktur
Tumorantigen, vorzugsweise maligitätsassoziiert, z. B.
HER2
EGFR
Trop2
CD-Antigene wie CD20, CD30, CD33
2. Antikörper fängt lösliche Wachstumsfaktoren ab
VEGF
EGFR-Liganden
2a. Antikörper blockiert die Rezeptoren für die löslichen Wachstumsfaktoren unter 2.
3. Antikörper hält Strukturen des Immunsystems auf „scharf“ geschaltet
Immunagonistische Antikörper – Checkpoinit-Inhibitoren, auch Immuncheckpoint-Inhibitoren (ICI)
CTLA4
PD-1
PD-L1
LAG3
Die Antikörper unter 1. können weiter eingeteilt werden in unkonjugierte, „nackte“ Antikörper und in toxingekoppelte Antikörper (ADC = Antibody Drug Conjugate). Hierbei dienen die Antikörper als „Schlepper“ für ein Toxin, das, würde man es ohne Antikörper geben, viel zu toxisch für den Patienten sein würde. Voraussetzung für solche ADCs ist, dass sie an einer tumorspezifischen Struktur binden können, im peripheren Blut stabil bleiben, nach Andocken an die Tumorzelle in diese internalisiert werden und erst in der Zielzelle gespalten werden, damit das Toxin dort selektiv wirken kann.
Merke:
Derzeit sind keine tumorexklusiven Antigene bekannt, nur eine mehr oder weniger tumorspezifische Überexpression (s. unter 1.).
Antikörper lassen sich auch mit Radionukliden koppeln. Diese werden als RIC = Radio Immuno Conjugate bezeichnet, haben aber eine therapeutisch untergeordnete Bedeutung. Mit RICs erfolgt sozusagen eine Bestrahlung von innen.
3.6.1 Immunagonistische Antikörper – Syn. Checkpoint-Inhibitoren, Immuncheckpoint-Inhibitoren (ICI)
Das menschliche Immunsystem kann vom Grundsatz her eine Tumorbildung verhindern. Tumorzellen haben allerdings die Fähigkeit, sich der Erkennung des Immunsystems zu entziehen. Dies geschieht u. a. über sog. inhibitorische Immunkontrollpunkte (Immuncheckpoints). Zum Selbstschutz des Wirtes (Patient) wird eine angestoßene Immunreaktion durch bestimmte Moleküle wieder gebremst. Mit neuen Medikamenten – bestimmte monoklonale Antikörper – versucht man die Inhibition zu umgehen und das Immunsystem nachhaltig auf „scharf“ gestellt zu lassen.
3.6.2 Zytotoxische T-Zellen und Immuncheckpoints
Eine wichtige Rolle bei der Tumorbekämpfung durch das Immunsystem spielen T-Zellen. Ihnen werden über ihren T-Zellrezeptor (TZR) permanent Antigene präsentiert. Da grundsätzlich alle möglichen Antigene präsentiert werden, muss die T-Zelle zwischen „fremd“ = böse und „eigen“ = gut = gesund unterscheiden können. Für das Signal „fremd“ oder „Gefahr“ werden kostimulatorische Moleküle benötigt, um die T-Zellen in Alarmbereitschaft zu versetzen.
Die Stärke und Dauer der T-Zellantwort wird durch kostimulatorische oder inhibitorische Signale ausbalanciert. Letzteres, um eine überschießende Immunantwort zu unterbinden und damit die Zerstörung von gesundem Gewebe zu verhindern. Das geschieht durch die sog. inhibierenden Checkpoint-Moleküle. Sie sind notwendig, um beispielsweise bei Infektionen, Kollateralschäden in der Peripherie zu vermeiden. Immuncheckpoints limitieren also eine dauerhafte Immunantwort.
Ein neues Phänomen, das bei der Therapie maligner Melanome mit Ipilimumab beobachtet wurde, ist das sog. „tumor flare“, auch als Pseudoprogression bezeichnet. Die aktivierten T-Zellen wandern in das Tumorgewebe ein, es schwillt dadurch an und es macht den Anschein einer Tumorvergrößerung. Eine Melanomtherapie mit Ipilimumab darf hinsichtlich ihres Erfolges daher frühestens nach 12 Wochen beurteilt werden, nicht vorzeitiger und auch nicht voreiliger. Es muss mit dem Patienten besprochen werden, dass seine Tumorherde initial größer werden können – ein Zeichen der Immunantwort.
3.6.3 PD-1 – Programmed Cell Death Protein 1 und deren Liganden PD-L1/-L2
Das Programmed Cell Death Protein[3] (PD-1) und seine Liganden (PD-L1 und PD-L2) stellen einen weiteren Immuncheckpoint dar. Physiologische Aufgabe des PD-1 ist auch wieder die Begrenzung von Kollateralschäden bei einer ansonsten überschießenden Immunreaktion und es dient der Wirtstoleranz (Erkennen von selbst, eigen). PD-1 ist in aktivierten T-Zellen während der Effektorphase heraufgeregelt. PD-1 hat 2 verwandte Liganden: PD-L1, auch B7-H1, und PD-L2, auch B7-DC. Während CTLA4 die T-Zellen bereits kurz nach deren Aktivierung bremst, wird durch PD die T-Zellantwort in den (peripheren) Geweben zur Zeit einer inflammatorischen Immunantwort gehemmt. Die Ausbildung von PD-1-Liganden (PD-1L) seitens des Tumors ist eine andere Art des „Tumor Escape“. Sie stellen im Tumor Microenvironment einen wesentlichen Resistenzmechanismus dar. Die Interaktion zwischen PD-1 und seinen Liganden reguliert die T-Zell-Aktivität herab. Diese führt zu einem Zustand der Anergie im Sinne einer Erschöpfung der antigenspezifischen T-Zellen. Der Tumor entzieht sich dem Immunsystem. Dieser Zustand kann durch eine PD-1-Blockade rückgängig gemacht werden. Anders ausgedrückt soll mit diesem Therapieprinzip die Immuntoleranz gegen den Tumor herabgesetzt werden. Er soll dem Immunsystem wieder sichtbar gemacht werden. Die anti PD-1 Antikörper sind PD-1-Antagonisten ohne intrinsische Aktivität (keine ADCC, CDC usw.). Die PD-1 Liganden können nicht binden, die T-Zelle bleibt aktiv.
Merke:
CTLA4 reguliert prädominant die T-Zell-Aktivierung.
PD-1 reguliert prädominant die Effektor-T-Zellaktivität in Geweben inkl. Tumoren.
Insbesondere auf soliden Tumoren ist das PD-L1 Antigen überexprimiert. Der Ligand führt durch Bindung an PD-1 ebendfalls zur T-Zellhemmung. Klinisch macht sich das ebenfalls als Immuntoleranz gegenüber dem Tumor bemerkbar. Anders ausgedrückt: Es liegt wieder eine (lokale) Immunosuppression durch den Tumor vor. Es konnte mittels Immunohistochemie nachgewiesen werden, dass das PD-L1 Antigen bei verschiedenen menschlichen Tumoren konstitutiv exprimiert wird. Das betrifft beispielsweise Tumore der Lunge, der Ovarien, des Kolons und bei Melanomen.
3.6.4 Anti-PD-1- und Anti-PD-1L-Antikörper
Mit Anti-PD1- und Anti-PD-1L-Antikörpern wird versucht, die periphere Tumortoleranz zu brechen. Die Entwicklung neuer Antikörper, neuer Kombinationen und Einsatzgebiete ist rasant.
PD-1 wird auf aktivierten T-Zellen während der Effektorphase hochreguliert. PD-L1 und PD-L2 auf den Tumorzellen belegen den PD-Rezeptor auf den T-Zellen und regeln deren Aktivität herunter. Die Tumorzellen werden sozusagen als „eigen“ toleriert. Es kommt zu Selbsttoleranz und damit zum sog. Tumor Escape. Durch Bindung von Antikörpern an PD-1 oder deren Liganden (PD-1L) kommt die beschriebene Interaktion nicht zustande und die T-Zelle bleibt aktiv. Der Tumor Escape wird unterbunden.
Merke:
Mit den unkonjugierten und konjugierten Antikörpern wird der Tumor direkt bekämpft.
Die ICIs (re-)aktivieren das Immunsystem (T-Zellen) und sind nicht direkt gegen die Tumorzellen aktiv.
3.6.5 LAG3-Antikörper
LAG3 = CD223 ist ein ko-inhibitorischer Rezeptor der T-Zellaktivierung und Zytokinsekretion. Die Aktivierung von LAG3 kann tatsächlich genutzt werden, um das Auftreten von Autoimmunerkrankungen zu verhindern. LAG3 findet sich auf Zellmembranen von TILs, aktivierten CD4+, CD8+, Tregs, NK-, B- und dendritischen Zellen. Es gibt einen großen Synergismus zusammen mit PD-1. So treibt eine hohe LAG3-Expression Tumorwachstum an. Man spricht auch von Immunosuppression im tumoralen Microenvironment und damit in der Folge von intrinsischer Resistenz. Durch die inhibitorischen Rezeptoren verlieren die T-Zellen ihre Effektorfunktionen. Es resultieren sog. exhausted T-Cells. Relatlimab ist in fixer Kombination mit Nivolumab (beide Antikörper in einem Vial) zur Erstlinienbehandlung des fortgeschrittenen, nicht resezierbaren oder metastasierten Melanoms bei Erwachsenen und Jugendlichen im Alter ab 12 Jahren zugelassen.
3.7 Spezielle Fragestellungen
3.7.1 ICI in Kombination mit CTX?
Nachdem der Stellenwert der ICI klar wurde, ergab sich schnell die Frage, ob man ICI mit der konventionellen, aber immunsuppressiven Chemotherapie (CTX) kombinieren kann. Und wenn ja, wie? Sequenziell? Zuerst die CTX gefolgt von ICI? Oder doch umgekehrt? Als First-in-Class Kombination wurde im September 2018 die Kombination Pem/Pem/Carbo = Pembrolizumab + Pemetrexed + Carboplatin gegen das nicht-plattenepitheloide NSCLC zugelassen. Es folgten weitere Zulassungen von Anti-PD-1- und Anti-PD-L1-Antikörpern in Kombination mit konventioneller CTX kurz darauf, diese lassen sich aber wegen der rasanten Dynamik der Zulassungserweiterungen hier nicht darstellen. Es wird auf die aktuellen Fachinformationen verwiesen.
3.7.2 Was ist mit der Kombination von ICI mit Glukokortikoiden?
Glukokortikoide sind bekanntermassen auch immunsuppressiv – in Abhängigkeit von Dauer und Dosis. Also ergab sich die Frage: wenn ICIs mit hoch emetogener CTX kombiniert werden, kann im Rahmen der Antiemese Dexamethason beibehalten werden? Klare Antwort: JA! Zum einen wurde im Rahmen der Zulassungsstudien Dexamethason leitliniengerecht beibehalten und der experimentelle Kombinationsarm aus ICI und hoch emetogener CTX war der Standard-CTX trotzdem überlegen. Zum anderen nahm sich die amerikanische Krebsgesellschaft des Themas an und hat 2020 eine entsprechende Leitlinie publiziert: „Antiemetics: ASCO Guideline Update“, im Journal of Clinical Oncology (https://ascopubs.org/doi/pdf/10.1200/JCO.20.01296, aufgerufen am 12.3.2021). Dort heißt es, es gibt keine klinische Evidenz, das Weglassen des Dexamethasons im Rahmen der hoch emetogenen Antiemese zu fordern.
Merke:
Immunagonistische ICI können mit immunsuppressiver CTX kombiniert werden.
Ist die CTX bei einer solchen Kombination hoch emetogen, verbleibt das Dexamethason im Prophylaxeregime gemäß den Antiemeseleitlinien.
Dass eine solche Kombination funktioniert, kann man sich mit den Eliminationshalbwertszeitender IC-Antikörper klarmachen. Eine Faustregel besagt, dass nach 5 Halbwertszeiten (HWZ) kein aktiver Wirkstoff mehr im System (im Patienten) vorhanden ist und damit auch keine Wirkung mehr. Bei den ICI liegt die 5. HWZ im Bereich von Monaten, wie die folgende Tabelle zeigt. Allerdings weiß bisher niemand, ob die immunagonistische Wirkung nach dem Absetzen eines IC-Antikörpers nach 5 HWZ wirklich zu Ende ist, oder noch länger andauert, da irAE auch Wochen bis Monate nach dem Absetzten auftreten können. Die potenziell immunsuppressive Wirkung von Dexamethason, sofern sie im Rahmen der Emeseprophylaxe überhaupt eine Rolle spielt, ist aber bei weitem nicht so langanhaltend.
| INN | Ziel | HWZ [d] → | 5 HWZs [d] → | 5 HWZs [Wochen] → | 5 HWZs [Monate]* |
| Atezolizumab | PD-L1 | 27 | 135 | 19,3 | 4,4 |
| Avelumab | PD-L1 | 6,1 | 30,5 | 4,4 | 1,0 |
| Cemiplimab | PD-1 | 19 | 95 | 13,6 | 3,14 |
| Dostarlimab | PD-1 | 25,4 | 127 | 18,1 | 4,2 |
| Durvalumab | PD-L1 | 18 | 90 | 12,9 | 2,9 |
| Ipilimumab | CTLA4 | 15,4 | 77 | 11,0 | 2,5 |
| Nivolumab | PD-1 | 25 | 125 | 17,9 | 4,1 |
| Pemrolizumab | PD-1 | 22 | 110 | 15,7 | 3,6 |
| Tislelizumab | PD-1 | 23,8 | 119 | 17 | 3,9 |
| Toripalimab | PD-1 | 18 | 90 | 12,9 | 2,9 |
| Tremelimumab | CTLA4 | 18,2 | 91 | 13 | 2,9 |
| HWZ basierend auf den Angaben in FI / FPI | |||||
| Start | 1 HWZ | 2 HWZ | 3 HWZ | 4 HWZ | 5 HWZ |
| 100% | 50% | 25% | 12,50% | 6,25% | 3,125% |
| * 1 Monat wird mit 30,4375 d berechnet gemäß (3 x 365 d + 1 x 366 d)/48 Monate | |||||
Mit den ICI verbunden sind aber auch immunvermittelte, neuartige Toxizitäten (irAEs – immune related Adverse Events). Diese gilt es zu kennen und zu erkennen, damit das Immunsystem seinen eigenen Wirt (Patient) nicht angreift und dadurch schädigt. Durch das therapeutisch (über-)aktivierte Immunsystem fällt die Selbsttoleranz gegenüber dem Wirt, sodass gesunde körpereigene Strukturen von den Immunzellen angegriffen werden. Auf derartige Nebenwirkungen müssen die Patienten hingewiesen und ggf. auch unterwiesen werden (siehe hierzu auch Teil Nebenwirkungen).
| Monoklonale Antikörper | Wirkstoff (INN) | Zielstruktur |
| Nicht konjugierte AK | Alemtuzumab* Amivantamab Atezolizumab Avelumab Bevacizumab Blinatumumab Cemiplimab Cetuximab Daratumumab Dinutuximab Dostarlimab Durvalumab Elotuzumab Elranatamab Epcoritamab Glofitamab Ipilimumab Isatuximab Margetuximab Mogamolizumab Mosunetuzumab Naxitamab Necitumumab (außer Handel) Nivolumab Nivolumab + Relatlimab Obinutuzumab Panitumumab Pembrolizumab Pertuzumab Ramucirumab Rituximab Tafasitamab Talquetamab Teclistamab Tislelizumab | CD52-Antigen (B- + T-Lymphozyten) EGFR x MET PD-L1 PD-L1 VEGF CD3 x CD19 (T- & B-Zellen) PD-1 EGF-Rezeptor CD38 Gangliosid GD2 PD-1 PD-L1 SLAMF7 CD3 x BCMA CD3 x CD20 CD3 x CD20 CTLA4 (T-Zellen) CD38 HER2-Rezeptor CCR4 = chemokine receptor type 4 Anti-CD3 x Anti-CD20 Gangliosid GD2 EGF-Receptor PD-1 PD-1 + LAG-3 im Gemisch CD20-Antigen (B-Lymphozyten) EGF-Rezeptor PD-1 HER2-Rezeptor Dimerisierungs-Inhibitor (HDI) VEGF-R2 CD20-Antigen (B-Lymphozyten) CD19 CD3 x GPRC5D CD3 x BCMA HER2-Rezeptor |
| Nicht konjugierte AK | Toripalimab Trastuzumab Tremelimumab | PD-1 PD-1 CTLA4 (T-Zellen) |
| Konjugierte AK (ITs) | Belantamab Mafodotin Brentuximab Vedotin Enfortumab Vedotin Gemtuzumab Ozogamicin Inotuzumab Ozogamicin Loncastuximab Tesirin Mirvetuximab Soravtansine Polatuzumab Vedotin Sacituzumab Govitecan Tisotumab Vedotin Trastuzumab Deruxtecan (DXd) Trastuzumab Emtansin (T-DM1) | BCMA (B Cell Maturation Antigen); Myelomzellen CD30, Hodgkin-Lymphomzellen Nectin 4 CD33-Antigen (AML-Zellen) CD22 (B-Zellen) CD19 (B-Zellen) FRα (Folat Rezeptor α) CD79b (B-Zellen) Trop-2 (Trophoblast cell-surface antigen-2) Tissue factor (TF) HER2-Rezeptor HER2-Rezeptor |
| Radioimmunkonjugate (RICs) | 90Y-Ibritumomab Tiuxetan | CD20-Antigen |
| Fusionsproteine | ||
| Nichtkonjugierte | Aflibercept | VEGF-A und -B |
| Tebentafusp | CD3 x TCR Steuerdomäne | |
| Konjugierte | Moxetumumab Pasudotox | CD22 (B-Zellen) |
| Tagraxofusp | CD123 (= α-Kette des IL-3 Rezeptors) | |
| * Keine onkologische Zulassung mehr. s.c.-Gabe war nie explizit zugelassen, wurde aber aus Gründen der besseren Verträglichkeit basierend auf einer Studie von Lundin et al. so gemacht (Blood 2002; 100: 768–773). | ||
3.8 Niedermolekulare Kinase-Inhibitoren (nmKI)
Eine neue Klasse von Zytostatika stellen niedermolekulare Hemmstoffe von Kinasen dar (-nibe). Kinasen sind Enzyme, die (reversibel) einen negativ geladenen Phosphatrest von ATP auf Funktionsproteine übertragen, wodurch oftmals eine Aktivierung erfolgt. Die Proteinkinasen bilden eine Superfamilie, die nach unterschiedlichen Kriterien in verschiedene Gruppen (17), Familien (134) und Unterfamilien (201) subdifferenziert werden können, nach der Art der modifizierten Gruppe – also nach der Aminosäure, auf das die Phosphatgruppe übertragen wird. So können Kinasen weiter unterteilt werden in Serin-/Threonin- oder Tyrosinkinasen. Die Serin-/Threoninproteinkinasen (Ser/Thr-PK) und die Tyrosinproteinkinase (Thr-PK) stellen 2 große Subgruppen innerhalb einer Familie dar. Nach der zellulären Lokalisation können die Kinasen unterteilt werden in:
- nicht-transmembrane Kinasen, auch „zytosolische“ Kinasen, die also gelöst in der Zelle herumschwimmen und
- transmembrane Kinasen, auch Rezeptorkinasen, die in der Zellwand lokalisiert sind und die Funktion eines Rezeptors haben (z. B. EGFR, VEGFR).
Störungen der Kinasefunktion (oft Überfunktion) sind an diversen Erkrankungen beteiligt, unter anderem auch bei der Entstehung und Aufrechterhaltung von Krebs. (Über-)aktivierte Kinasen schützen die Tumorzelle vor dem physiologischen, programmierten Zelltod (Apoptose), haben also eine antiapoptotische Wirkung. Die Apoptose ist ein für das Individuum schmerzfreier Vorgang des natürlichen Zellumsatzes und sozusagen das Gegenteil einer Nekrose (z. B. Verbrennung). Kinase-Inhibitoren wirken durch eine Blockade der ATP-Bindungsstelle. Daher werden sie auch als ATP-Mimetika oder als „small molecular kinase inhibitor“(smKI), deutsch nmKI – niedermolekulare Kinase-Inhibitoren, bezeichnet. Das Wirkprinzip eines nmKI am Beispiel einer zytosolischen Kinase ist in der Abbildung unterhalb wiedergegeben und verdeutlicht die Angriffspunkte von einem nmKI und einem moAK an einer Rezeptortyrosinkinase.
Die ATP-Bindungsstelle ist in der Natur weit verbreitet (Glykolyse, Zitratzyklus), womit sich die Spezifität dieser Substanzen wieder relativiert. Werden mehr als nur die eigentlich gewünschten Kinasen gehemmt (Normalvarianten oder sog. Bystander-Kinasen), kann das ein Grund für unerwünschte Arzneimittelnebenwirkungen sein (siehe Abschnitt Targeted Therapies). Der überwiegende Teil der derzeit im Einsatz befindlichen smKI wird oral verabreicht. Zur Benennung der Wirkstoffe siehe Abschnitt Namensgebung.
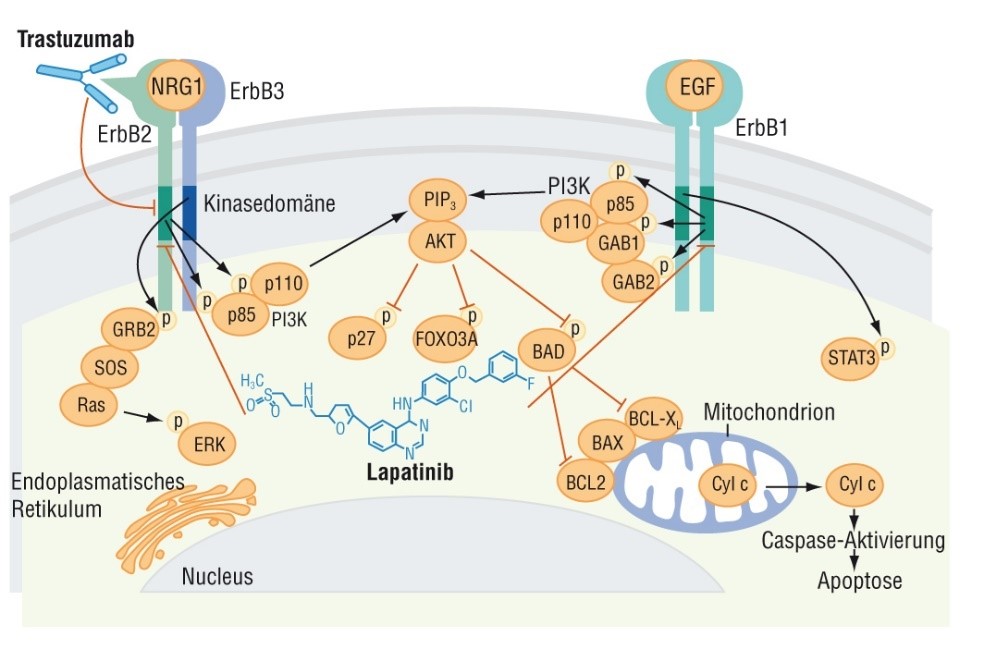
| Niedermolekulare Kinase-Inhibitoren (nmKI) | |
| Wirkstoff (INN) | Zielkinase(n) |
| Acalabrutinib | BTK |
| Afatinib | ErbB1, ErbB2, ErbB4 |
| Alectinib | ALK |
| Alpelisib | PI3Ka |
| Asciminib | Abl/BCR-Abl, mBCR/Abl inkl. T315I-Abl |
| Avapritinib | PDGFRA, PDGFRA D842 Mutanten, multiple KIT exon 11, 11/17 und 17 Mutanten, wild Typ KIT, PDGFRB, CSFR1 |
| Axitinib | VEGFR-1, 2, 3, PDGF-R, c-kit |
| Binimetinib | MEK 1, MEK 2 |
| Bosutinib | BCR/Abl, Src |
| Brigatinib | ALK, ROS1, IGF-1R, FLT-3, EGFR deletion and point mutations |
| Cabozantinib | RET, MET, VEGFR-1, -2, -3, KIT, TRKB, FLT-3, AXL, TIE-2 |
| Capiversitib | AKT |
| Capmatinib | MET inkl. Mutationen mit Exon 14 skipping |
| Ceritinib | ALK, IGF-1R, InsR, ROS1 |
| Cobimetinib | MEK 1, MEK 2 |
| Copanlisib1 | PI3 alpha/delta |
| Crizotinib | ALK, HGFR, c-Met, RON |
| Dabrafrenib | B-RAFV600E, B-RAFV600K, B-RAFV600D, wild-type B-RAF, C-RAF, SIK1, NEK11, LIMK1 |
| Dacomitinib | EGFR family (EGFR/HER1, HER2, HER4), DDR1, EPHA6, LCK, DDR2, MNK1 |
| Dasatinib | BCR/Abl, Src-Familie (Src, Lyn, Lck, Hck, Yes, Fyn), c-Kit, PDGF-R-β, RAF, EPHA2 |
| Duvelisib2 | PI3Kd + g |
| Encorafenib | B-RAFV600E, B-RAFV600K, wild-type B-RAF, C-RAF |
| Entrectinib | TRKA, TRKB, TRKC, ROS1, ALK, JAK2, TNK2 = ACK1 |
| Erdafitinib | mFGFR2, -3 (FGFR1, FGFR2, FGFR3, FGFR4 RET, CSF1R, PDGFRA, PDGFRB, FLT4, KIT, VEGFR2) |
| Erlotinib | ErbB1 |
| Everolimus | mTOR |
| Fedratinib | JAK 2 (-1, -3), FLT3ITD |
| Fruquintinib | VEGFR 1-3 |
| Futibatinib | FGFR |
| Gefitinib | ErbB1, (ErbB2?) |
| Gilteritinib | FLT3 |
| Ibrutinib | BTK |
| Idelalisib | PI3 delta |
| Imatinib | BCR-ABL, c-Kit, PDGF-R |
| Lapatinib | ErbB1, ErbB2 |
| Larotrectinib | TRK A-C |
| Lenvatinib | VEGFR-1, 2, 3, PDGF-R α, FGFR-1-4, RET, Kit |
| Lorlatinib | ALK, ROS1, TYK1, FER, FPS, TRKA, TRKB, TRKC, FAK, FAK2, ACK |
| Midostaurin | wtFLT3, FLT3 Mutantenkinasen ITD und TKD KIT Wildtyp und D816V Mutante, PDGFRα/β VEGFR2, PKC-Familie (Protein Kinase C), c-Kit PDGF-R |
| Momelotinib | wild type JAK1/2, mutant JAK2V617F |
| Neratinib | HER2/HER4 |
| Nilotinib | BCR/Abl, c-Kit, PDGF-R |
| Nintedanib | VEGFR-1, 2, 3, PDGF-R α und -β, FGFR-1-3 |
| Osimertinib | EGFR T790M |
| Pazopanib | VEGFR- (-1 bis -3), PDGFR-α und -β, FGFR-1 und -3, Kit, Itk, Lck, c-Fms |
| Pemigatinib | FGFR2 Fusion oder Rearrangement |
| Pexidartinib | CSF1R, (KIT, FLT3-ITD) |
| Pirtobrutinib | BTK |
| Ponatinib | Abl, T315I-Abl, VEGF-R, PDGF-R, FGFR, EPH Rezeptor, SRC Familie, KIT, RET, TIE2, FLT3 |
| Pralsetinib | RET (fusionspositiv) |
| Quizartinib | FLT3-ITD |
| Regorafenib (außer Handel, gezogen aber importierbar) | RET, VEGFR-1, -2, 3, KIT, PDGFR–α und -β, FGFR-1 und -2, TIE2, DDR2, Trk2A, Eph2A, RAF-1, B-RAF, B-RAFV600E, SAPK2, PTK5, Abl |
| Reprotrectinib | ROS-1, TRK A-C |
| Ripretinib | KIT, PDGFRa, (PDGFRB, TIE2, VEGFR2, and BRAF) |
| Ruxolitinib | JAK 1/2 (-1, -3) |
| Selpercatinib | RET (fusionspositiv) |
| Selumetinib | MEK 1/2 |
| Sorafenib | RAF (C-RAF, B-RAF, Raf-1), VEGF-R (-2 + -3), PDGF-R-β, c-KIT, FLT-3 |
| Sunitinib | c-Kit, VEGF-R (-1 bis -3), PDGF-R, RET, FLT-3 |
| Temsirolimus | mTOR |
| Tepotinib | MET inkl exon 14 skipping |
| Tivozanib | c-Kit, VEGF-R (-1 bis -3), PDGF-R, RET, FLT-3 |
| Trametinib | MEK 1, MEK 2, B-RAFV600E |
| Tucatinib | HER2 (in vitro HER3, PAN HER?) |
| Vandetanib | VEGFR2, RET, BRK, TIE2, EPH, EGFR, Src-Familie |
| Vemurafenib | B-RAFV600E, C-RAF, A-RAF, wild-type B-RAF, SRMS, ACK1, MAP4K5, FGR |
| Zanubrutinib | BTK |
| 1 War nur in den USA zugelassen. Im November 2023 freiwillig vom Markt gezogen, da es in Verbindung mit einer Chemoimmuntherapie zu keiner klinischen Verbesserung kam. 2 Duvelisib ist mir schweren Nebenwirkungen belastet und erfordert eine sorgfältiges Nebenwirkungsmanagement. Der pharmazeutische Unternehmer hat entschieden, Duvelisib für die beiden zugelassenen Indikationen nicht auf dem deutschen Markt einzuführen. | |
3.9 Weitere therapeutische Angriffspunkte – Regulatorproteine und weitere Kinasen
Nach den klassischen Zytostatika, den monoklonalen Antikörpern und den Kinase-Inhibitoren (bis Anfang der 2000er-Jahre) sind weitere antitumorale Wirkstoffklassen erfunden worden, die im Folgenden z. T. gruppiert beschrieben werden. So handelt es sich bei den CDK4/6-Inhibitoren zwar auch um Hemmstoffe von Kinasen (Ser/Thr-Kinasen), jedoch um eine besondere Gruppe im Zellzyklus, die auch als solche vorgestellt wird. Die folgenden Substanzen und Substanzklassen wurden nach 2010 zugelassen.
3.9.1 Hemmstoffe Cyclin-abhängiger Kinasen (CDKi)
Cyclin-abhängige Kinasen (engl. cyclin-dependent kinases, CDKs) sind eine Familie von Proteinkinasen, die sowohl bei der Transkription als auch bei der Kontrolle des Zellzyklus eine Rolle spielen. Beim Menschen existieren 20 verschiedene CDKs. CDKs bilden Komplexe mit den regulatorischen, kurzlebigen Cyclinen, die zellzyklusabhängige Konzentrationsänderungen durchlaufen. Die Cyclin-abhängigen Kinasen sind nur in Verbindung mit ihrem zugehörigen Cyclin aktiv; m. a. W. das Cyclin verleiht ihnen erst Kinaseaktivität. Dadurch kann die Aktivität der CDKs in der Zelle zeitlich gesteuert und reguliert werden. So wird ermöglicht, dass sie ihre Funktion in bestimmten Phasen des Zellzyklus ausführen, die an die zyklischen Konzentrationsänderungen der Cycline gekoppelt sind. Binden beispielsweise Cyclin D1 und die Enzyme CDK4 und 6 aneinander, erhält die Zelle ein „gehe weiter“-Signal, um von einer Phase des Zellzyklus in die nächste überzutreten. Aufgrund der essentiellen Rolle, die Cyclin-abhängige Kinasen bei der Steuerung des Zellzyklus und somit bei der Proliferation von Zellen spielen, werden verschiedene Substanzen, die CDKs hemmen (sogenannte CDK-Inhibitoren), als Krebstherapeutika eingesetzt. Der erste Vertreter der Wirkstoffgruppe der CDK-Inhibitoren war Palbociclib, ein CDK4/6-Inhibitor. CDK4/6 kontrollieren den Übergang einer Zelle von der G1-Phase in die S-Phase und schaffen damit die Voraussetzung für eine Zellteilung. Durch eine Hemmung von CDK4/6 werden folglich die Zellteilung und damit das Tumorwachstum gehemmt.
Am Übergang der G1/S-Phase des Zellzyklus gibt es einen Restriktionspunkt, der das Fortschreiten des Zyklus zunächst verhindert. Hier ist der Transkriptionsfaktor E2F an das Retinoblastomprotein (Rb) gebunden und inaktiv. Wenn über verschiedene Signale das kurzlebige Cyclin D gebildet wird und die CDK4/6 daran binden, erfolgt eine Phosphorylierung von Rb und E2F wird freigesetzt. Aktive E2F-Proteine führen zur Transkription von Genen der DNS-Synthese, die während der S-Phase benötigt werden. Der Zellzyklus schreitet fort. Bei Tumoren, wie z. B. das Estrogenrezeptor positive, HER2-negative Mammakarzinom gilt die Überexpression von CDK4/6 als Schlüsselfaktor für die Resistenzbildung gegen eine endokrine Therapie (Aromatasehemmer ± LHRH-Agonist). Die Hemmung von CDK 4/6 ist der Angriffspunkt der Ciclibe. Die endokrine Therapie wirkt wieder.
| Hemmstoffe Cyclin-abhängiger Kinasen- CDK4/6 (Ser-/Thr-Kinasen) | Abemaciclib Palbociclib Ribociclib |
3.9.2 Hemmstoffe des Hedgehog-Signalpfads (SMO)
Hedgehog-Proteine sind Signalproteine, die eine wichtige Funktion für Zellwachstum und Differenzierung im Rahmen der Embryogenese haben. Der Signalweg ist nach seinem Liganden Hedgehog (Hh) benannt. Der Hh-Signalweg trat in der Evolutionsgeschichte sehr früh auf. Beim adulten Organismus ist der Shh-Signalweg wichtig für die Regulation der Zellteilung der adulten Stammzellen. Eine Fehlfunktion dieses Signalwegs führt zu massiven Fehlbildungen im Laufe der Embryonalentwicklung und kann bei Erwachsenen Karzinome verursachen. Wie häufig ist im Rahmen von Tumorerkrankungen der Signalweg aktiviert, obwohl keine Liganden an den Rezeptor binden.
Hh-Signalweg-Inhibitoren binden an den Smoothened Rezeptor (SMO), weshalb man auch manchmal die Bezeichnung SMO-Inhibitoren findet (SMOi). Als erste Substanz wurde 2013 Vismodegib zur Behandlung bestimmter Basalzellkarzinome zugelassen. Es folgte Sonidegib im Jahre 2015 mit ähnlicher Indikation. Seit 2020 ist Glasdegib in Kombination mit niedrig dosiertem Cytarabin (Cytarabine) für die Behandlung von neu diagnostizierter de novo oder sekundärer AML bei Erwachsenen, die nicht für eine Standard-Induktionschemotherapie infrage kommen, zugelassen.
| Hemmstoffe des Hegehog Signalwegs / SMOi | Glasdegib Sonidegib Vismodegib |
3.9.3 Hemmstoffe der Poly(ADP-ribose)-Polymerase (PARP-Inhibitoren)
DNS-Schäden, wie sie typischerweise durch alkylierende Zytostatika entstehen, können von Zellen über die „homologe Rekombination“ (HR) oder die Basen-Exzisionsreparatur (Base Excision Repair, BER) repariert werden. Das nukleäre Enzym PARP (derzeit 17 bekanne Isoenzyme) ist für die Reparatur von Einzelstrangbrüchen der DNS verantwortlich. PARP-Inhibitoren binden an das aktive Zentrum der PARPs, die mit der DNS assoziiert sind, und verhindern so deren Dissoziation („PARP Trapping“). Die PARP–DNS Addukte blockieren sterisch den Platz für die BER-Enzyme. Bei gehemmter PARP werden diese Einzelstrangbrüche nicht mehr repariert. Bei der nächsten Zellteilung kommt es dann zur Bildung von Doppelstrangbrüchen, wenn die Replikationsgabeln (siehe Abschnitt Wirkungs- und Resistenzmechanismen) auf das PARP-DNS-Addukt stoßen. Diese können normalerweise durch HR oder BER repariert werden und ermöglichen damit der Zelle doch noch das Überleben. In Zellen mit gestörter Doppelstrangreparatur, z.B. bei Patientinnen mit BRCA-1/2-Mutationen oder HR Defizienz, werden die Strangbrüche dann nur noch fehlerhaft oder fehleranfällig, wie z. B. wie mittels nicht-homologer Endverknüpfung (NHEJ), oder gar nicht mehr repariert. Die Folge davon ist, dass betroffene Tumorzellen in die Apoptose gehen. Tumoren mit einer Keimbahn- oder somatischen Mutation in den BRCA-Genen reagieren deshalb besonders empfindlich auf PARP-Inhibitoren. Da die gesunden Zellen BRCA-assoziierter Tumorpatientinnen nur den heterozygoten HR tragen, tolerieren sie die pharmakologische PARP-Hemmung gut. Da PARP auch eine Rolle bei der Hämatopoese, insbesondere der Erythropoese spielt, ist mit der Anwendung der PARPi eine gewisse Hämatotoxizität verbunden. Diese zeigt sich, substanzabhängig, in Anämie, Thrombozytopenie und Neutropenie.
| Hemmstoffe der Poly-(ADP-ribose)-Polymerase, PARP-Inhibitoren – PARPi | Niraparib Olaparib Rucaparib Talazoparib |
3.9.4 Hemmstoffe von Regulatorproteinen – Bcl-2
Die Apoptose – der nicht-nekrotische oder programmierte Zelltod, ist ein hochgradig geregeltes „Suizidprogramm“ der Zelle. Dieses Programm wird über pro- und antiapoptotische Faktoren geregelt. Dazu gehören Proteine der Bcl-2-Familie.
| Proteine der Bcl-2-Familie | |
| Antiapoptotische Wirkung | Proapoptotische Wirkung |
| Bcl-2 | Bad |
| Bcl-xL | Bax |
| Bak | |
| Bcl-xS | |
| Bik | |
| Bim | |
| Bid | |
Überaktivierungen von Bcl-2 führen zu einem Ungleichgewicht im Sinne einer Apoptoseresistenz und damit zur Proliferation von Gewebe und damit zu Tumoren. Proapoptotische Proteine sind auf Mitochondrien durch Bcl-2 gebunden. Wird Bcl-2 freigesetzt, kommt es zur Porenbildung auf den Mitochondrien (MOMP: Mitochondrial Outer Membrane Permeabilisation), zur Freisetzung von Cytochom C, einem starken Zellsignal, die Apoptose zu beginnen, der Bildung des sog. Apoptosoms, einem Enzymkomplex, sowie weiteren, letalen Effektormolekülen.
Mit Venetoclax steht der erste Anti-Bcl-2-Wirkstoff zur Verfügung. Er verursacht eine Verdrängung der antiapoptotischen Proteine und damit eine Verschiebung des Gleichgewichts in Richtung Apoptose mit den oben beschriebenen Vorgängen.
3.9.5 Hemmstoffe der Isocitratdehydrogenase (IDH)
Mutationen in den Genen IDH1 und IDH2 (IDH, Isocitratdehydrogenase) kommen bei Patienten mit
- Gliomen
- AML (akute myeloische Leukämie, bis zu 10%)
- MDS (myelodysplastisches Syndrom)
- angioimmunoblastischem T-Zell-Lymphom
- Chondrosarkom
- cholangiozellulärem Karzinom
und anderen Tumorarten vor.
Physiologisch spielt die IDH eine wichtige Rolle im Zitronensäurezyklus. Durch Mutationen im IDH 1/2 Gen kommt es zu einem Funktionsgewinn. Die IDH macht aus Isocitrat nicht mehr α-Ketoglutarat (α-KG), sondern 2-Hydroxyglutarat (2-HG). 2-HG übt epigenetische Effekte aus. 2-HG gilt als Onkometabolit und bewirkt unter anderem eine genomweite Histon- und DNS-Methylierung mit der Konsequenz der Stilllegung von Tumorsupressorgenen und Verhinderung der (funktionalen) Ausdifferenzierung. Durch die Akkumulation von 2-HG und der daraus resultierenden erhöhten 2-HG/α-KG-Ratio werden die epigenetischen Funktionen α-KG-abhängiger Enzyme (u.a. TET2, JMJD Histondemethylasen) gehemmt und ist mit einem ungünstigem Krankheitsverlauf assoziiert. Die IDH-Hemmung führt zu weniger 2-HG und zur Reduktion myeloischer Blasten und zur verbesserten und vermehrten Ausdifferenzierung myeloischer Zellen.
| Hemmstoffe der Isocitratdehydrogenase (IDH) | |
| IDH-1 | Ivosidenib |
| IDH-2 | Enasidenib |
3.9.6 XPO = CRM1
Bei Tumoren kann das Chromosome Region Maintenance 1 Protein (CRM1) = Exportin 1 oder XPO1 überexprimiert vorliegen. XPO mediiert den Export von Proteinen aus dem Zellkern, so z. B.
- Tumorsuppressorproteine (TSPs), inklusive p53, p21, BRCA1/2, pRB, FOXO sowie die
- mRNS onkogener Proteine und anderer Wachstumsregulatorproteine.
Der Export von TSPs aus dem Zellkern fördert das ungebremste Zellwachstum. Durch die Blockade von XPO werden die TSPs im Zellkern retiniert und endogene Tumorsuppressorprozesse restauriert. Mit anderen Worten: TSPs akkumulieren wieder im Zellkern, der Gehalt an Onkoproteinen wie c-myc und Cyclin D1 wird reduziert. Es kommt zum Zellzyklusarrest, gefolgt von der Apoptose.
Selinexor ist der 1st-in-Class XPO-Inhibitor, was zu der Wirkstoffklassenbezeichnung SINE geführt hat – Selective Inhibition of Nuclear Export.
3.9.7 GTPase Hemmstoffe
GTPasen sind eine große Familie von Hydrolaseenzymen, die an das Nukleotid Guanosintriphosphat (GTP) binden und es zu Guanosindiphosphat (GDP) hydrolysieren. GTPasen fungieren als molekulare Schalter oder Zeitgeber in vielen grundlegenden zellulären Prozessen.
Beispiele für diese Rollen sind:
- Signaltransduktion als Reaktion auf die Aktivierung von Zelloberflächenrezeptoren, einschließlich Transmembranrezeptoren wie solchen, die Geschmack, Geruch und Sehvermögen vermitteln.
- Proteinbiosynthese (auch bekannt als Translation) am Ribosom.
- Regulierung der Zelldifferenzierung, -proliferation, -teilung und -bewegung.
- Translokation von Proteinen durch Membranen.
- Transport von Vesikeln innerhalb der Zelle und vesikelvermittelte Sekretion und Aufnahme durch GTPase-Kontrolle des Aufbaus der Vesikelhülle.
GTPasen sind aktiv, wenn sie an GTP gebunden sind, und inaktiv, wenn sie an GDP gebunden sind.
In der Onkologie bekannte GTPasen sind K-RAS und N-RAS. Kommt es zu einer Mutation im für K-RAS kodierenden Gen, ist häufig ein Kontrollverlust des betreffenden Proteins die Folge. In diesem Falle kommt es dann zu unkontrollierbaren und zahlreich ablaufenden Zellteilungen. Seine wichtige Beteiligung an der Entstehung oder der Chemotherapieresistenz von Tumoren macht K-RAS zu einem wichtigen Angriffspunkt in der Entwicklung neuartiger Krebsmedikamente. So muss vor dem Einsatz der Anti-EGFR-Antikörper Cetuxi- und Panitumumab auf pan-RAS-Wildtyp getestet werden, weil deren Einsatz bei mutiertem RAS sinnlos ist (mutiertes K-RAS „feuert“ in der Signaltransduktionskette unterhalb von EGFR). K-RAS galt lange Zeit als „undruggable“. Nun gibt es derweil 2 K-RAS-Inhibitoren, aber nur gegen die K-RAS-G12C-Mutation.
| Hemmstoffe von K-RAS (nur G12C Mutation) | Adagrasib |
| Sotorasib |
3.9.8 Notch Pathway und Gamma Sekretase
Die γ-Sekretase ist ein aus mehreren Untereinheiten bestehendes Enzymkomplex und hat verschiedene biokatalysatorische Funktionen. Insbesondere bei der Entwicklung von Geweben und Organen ist sie essenziell. Der Komplex besteht aus mehreren Untereinheiten mit den Protease-Untereinheiten
- Präsenilin-1- oder -2-Homodimer
- Nicastrin
- Stabilisationfaktor APH-1 und
- Präsenilin-Enhancer 2 (PEN-2).
Diese schneiden Transmembranproteine innerhalb der Transmembrandomäne. Die γ-Sekretase schneidet das Amyloid-Precursor-Protein (APP, aggregiert als „falsch gefaltetes“ β-Amyloid bei Alzheimer-Erkrankungen), ein großes integrales Membranprotein, nach der β-Sekretase. Darüber hinaus wird das Rezeptorprotein Notch geschnitten.
Der Notch-Rezeptor bindet den membranständigen Liganden Delta von Oberflächen anderer Zellen. Durch seine folgende Proteolyse durch die γ-Sekretase wird der Notch-Signalweg aktiviert.
Der Notch-Rezeptor wird dreimal proteolytisch geschnitten, das erste Mal im Golgi-Apparat und danach zwei weitere Male, nachdem Delta gebunden hat. Zuerst wird die extrazelluläre Domäne (EC) von der ADAM-Metalloprotease (A Disintegrin And Metalloproteinase) nahe der Plasmamembran abgespalten. Dann wird der zytoplasmatische Teil des membranständigen Notch-Rezeptors (IC) – vermutlich durch Präsenilin-1 – abgetrennt und kann nun frei in den Zellkern diffundieren, wo das Fragment in einem Komplex, zusammen mit anderen Regulatorproteinen, bindet und so die Expression von Notch-Response-Genen reguliert. Chromosomale Translokationen, Punktmutationen und chromosomale Amplifikationen führen zur konstitutiven Aktivierung des Notch-Signalwegs und so zu Tumoren. Die Notch-Signalgebung kann durch Hemmung der γ-Sekretase inhibiert werden.
Die Notch-Signalgebung ist überaktiv bei Desmoidtumoren, auch als aggressive Fibromatosen bezeichnet. Sie entstehen aus den Zellen, die die Bindegewebestruktur des Körpers ausmachen, sind nicht metastasierend, wachsen aber in umliegende Gewebe oder Strukturen hinein und zerstören diese. Sie können überall im Körper auftreten, sind aber häufiger in bestimmten Bereichen wie der Bauchwand, im Bauchraum oder in den Beinen und Armen. Die Symptome können je nach Standort variieren und umfassen:
- Schmerzen
- eine sichtbare Masse oder Schwellung
- Funktionsstörungen des betroffenen Bereichs und
- Fälle von intestinale Obstruktion oder andere Komplikationen.
Bisherige therapeutische Optionen bestanden aus einer Kombination aus Chirurgie, Medikamenten, Strahlentherapie oder Active Surveillance. Desmoidtumore sind schwer zu behandeln und haben ein hohes Rezidivrisiko, auch nach einer scheinbar erfolgreichen Behandlung. Mit Nirogacestat steht nun der erste γ-Sekretase-Inhibitor zur Verfügung, mit dem auf die maligne Notch-Signalgebung Einfluss genommen werden kann. Hinweis: Nirogacestat ist ovartoxisch im Sinne von fruchtbarkeits- und fortpflanzungsschädlich.
3.9.9 Hemmstoffe der Ornithindecarboxylase
Polyamine, geradkettige Amine, die biosynthetisch von Aminosäuren abstammen, kontrollieren Genexpression auf der transkriptionalen, posttranskriptionalen und translationalen Ebene. Multiple, zelluläre onkogene Signalwege sind in der Regulation der Transkription und Translation von Polyamin-metabolisierenden Enzymen beteiligt.
Genetische Alterationen, Expressionslevel und Aktivität von Polyamin-metabolisierenden Enzymen verändern sich schnell während der Tumorgenese. Die Folge ist: Man findet hohe Polyaminspiegel in vielen menschlichen epithelialen Tumoren.
N-Myc, kurz für N-Myc-Protoonkogen-Protein, ist ein Transkriptionsfaktor der MYC-Familie. N-Myc wird in frühen Stadien der Embryonalentwicklung von neuronalen Progenitorzellen exprimiert. Im Gegensatz zu C-Myc wird die Genexpression von N-Myc eigentlich nach der Zelldifferenzierung abgeschaltet. Er reguliert die Transkription verschiedener Gene in Tumorzellen, insbesondere in Tumoren des Nervensystems. Die Expression von N-Myc ist bei Tumoren des Nervengewebes hochreguliert. Insbesondere bei Neuroblastomen und Medulloblastomen, aber auch bei der akuten myeloischen Leukämie gilt N-Myc als Indikator für einen progressiven Verlauf und eine schlechte Prognose. Eine Inhibition der Ornithindecarboxylase, die Ornithin zu Tetramethylendiamin (1,4-Diaminobutan) decarboxyliert, restauriert das Ungleichgewicht im sog. LIN28/Let-7 metabolic pathway.
Eflornithin ist ein Ornithindecarboxylasehemmstoff, für Erwachsene und Kinder mit Hochrisiko-Neuroblastom, nach mindestens partieller Remission unter vorheriger, multimodaler Therapie inkl. Anti-GD2-Immuntherapie zugelassen ist. Hinweis: Eflornithin ist keineswegs neu. Es ist in diversen Dermatika zur Behandlung eines Hirsutismus im Gesicht bei Frauen enthalten.
3.10 Weitere Immunonkologika – nach ICIs
3.10.1 Hämatologie in Fahrt – von CAR-T-Zellen und TRUCKs, the living drugs
Die CAR-T-Zelltechnologie stellt ein anspruchsvolles Verfahren im Rahmen der Immunonkologie dar. CAR steht für chimäre Antigenrezeptor T-Zellen. Es handelt sich dabei um eine zelluläre Immuntherapie auf patientenindividueller Basis. Diese Therapeutika fallen unter den Begriff der ATMPs = Advanced Therapy Medicinal Products. Die deutsche Terminologie lautet Arzneimittel für neuartige Therapien.
Die ATMPs umfassen Gentherapeutika, somatische Zelltherapeutika und biotechnologisch bearbeitete Gewebeprodukte, sogenannte TEP – Tissue Engineered Products – also modifizierte, (lebende) Zellen und andere Gewebe. Kombinierte ATMP, also Kombinationen von ATMP und Medizinprodukten, zählen auch zu den ATMP. Sie sind besonderen Qualitätsnormen und Zulassungsverfahren unterworfen.
3.10.2 Verfahren und Wirkprinzip
Dem Patienten muss zunächst eine ausreichende Menge an T-Zellen entnommen werden. Dies geschieht durch eine nicht-stimulierte Leukapharese (ohne G-CSF). Die separierten, patienteneigenen T-Zellen werden dann ex vivo, d.h. außerhalb des Körpers, auf genetischer Ebene mit spezifischen, T-Zell aktivierenden, chimären Antigenrezeptoren (CARs) ausgestattet. Dies geschieht mit Hilfe lenti- oder retroviraler Gentransfervektoren, welche die genetische Information für den CAR auf die T-Zellen übertragen. Diese werden stabil in das Genom der T-Zellen eingebaut. Somit wird bei Aktivierung und Teilung der T-Zellen die genetische Information für den CAR an die Tochterzellen weitergegeben.
CARs setzen sich aus einer extrazellulären Bindedomäne, einer Linkerregion (Hinge), einer Transmembrandomäne und einer intrazellulären Signalsequenz zusammen. Für das Erkennen der Tumorzellen ist die Bindedomäne zuständig. Sie besteht aus einem Antikörperfragment (scFv – single chain variable Fragment), das an ein möglichst ausschließlich auf Tumorzellen vorhandenes Oberflächenantigen bindet. Das scFv ist derzeit murinen Ursprungs, daher auch chimärer Antigenrezeptor. Die Transmembrandomäne sorgt für eine Verankerung und Präsentation des CARs auf der Oberfläche der T-Zellen. Die zweite für die therapeutische Aktivität entscheidende Komponente ist die Signalsequenz, die nach Bindung des CARs an Tumorzellen für eine Aktivierung der T-Zellen sorgt. CAR-Ts der ersten Generation hatten nur eine, aber essenzielle, Aktivierungsdomäne (CD3z). Diese Erstgenerationskonstrukte hatten aber eine limitierte Aktivierungs- und Expansionskapazität. Daraufhin wurden kostimulatorische Moleküle (CD28, 4-1BB) als Tandemkonstrukte inkorporiert. Die Bindung an das Tumorantigen via scFv kann MHC-unabhängig erfolgen.
Merke:
Chimäre Antigen-Rezeptoren (CAR) sind synthetische, tumorspezifische Rezeptoren, die in vitro genetisch reprogrammiert sind, um unter Verwendung der patienteneigenen Lymphozyten MHC-unabhängig an Tumorantigene zu binden und T-Zellen in die Lage zu versetzen, antigenexprimierende Tumorzellen zu erkennen und zu vernichten.
Durch Kombination mehrerer Signalsequenzen kann die Aktivierung der CAR-T-Zellen nach Tumorbindung als auch die daraus resultierende Zytokinproduktion und Proliferation verstärkt werden (3.-generations CAR-Ts). Das kostimulatorische Molekül CD28 stimuliert den B7 Signalweg (siehe Abschnitt Zytotoxische T-Zellen), während 1-4BB den Tumornekrosefaktor assoziierten Faktorensignalpfad triggert. CAR-T-Zellen mit CD28 expandieren schnell, wohingegen solche mit 1-4BB langsamer proliferieren, was sich auch im Toxizitätsprofil widerspiegelt (siehe CAR-T-Zellen assoziierte Toxizität). CAR-T-Zellen mit 1-4BB persitieren auch länger im Patienten. Weitere Unterschiede in den Phänotypen sind:
CAR-T-Zellen mit 1-4BB haben
- Eine höhere Proliferationsrate, nicht Geschwindigkeit, s.o.
- Eine stärke Differenzierung in zentrale Gedächtniszellen (Central Memory T-Cells)
- Eine Steigerung der intrazellulären mitochondrialen Funktion der CD8-positiven Zellen jeweils im Vergleich mit CD28.
Es ist eine absehbare Konsequenz, dass CAR-T-Zellen der 3. Generation entwickelt werden, die sowohl CD28 als auch 4–1BB oder weitere Sequenzen (OX40) als kombinierte Kostimulation enthalten werden.
Die Entwicklung genmodifizierter CAR-T-Zellen geht permanent weiter. Zur Toxizitätsverminderung und um gesundes Gewebe vor dem Angriff von CAR-T-Zellen zu verschonen, arbeitet man an sog. Tandem CAR-Ts, die erst dann „scharf gestellt“ werden, wenn zwei tumorspezifische Antigene gleichzeitig vorliegen. Ein anderes Konzept sind sog. iCARs. Trifft eine solche T-Zelle auf nicht-tumoröses Normalgewebe, wird ein inhibitorisches Signal ausgelöst und die T-Zelle nicht aktiviert.
3.10.3 Das TRUCK-Konzept
Die verfügbaren CAR-T-Produkte haben ihre Anwendung bei B-Zell-Neoplasien. Der Erfolg in der Hämatologie kann bis dato nicht auf solide Tumore übertragen werden. Grund dafür ist ein zu immunsuppressives TME dieser Tumore, welches neben immunsuppressiven Zytokienen auch eingewanderte/angelockte imminsuppressive Tregs und MDSC enthält. Dieses repressive Milieu versucht man mittels des TRUCK-Konzepts zu umgehen. Anders ausgedückt: Man versucht die insuffiziente Produktion proinflammatorischer Zytokine der im Tumormilieu akkumulierenden T-Zellen zu verstärken. TRUCK steht für T-cells redirected for antigen-unrestricted cytokine-initiated killing. Es handelt sich bei den TRUCKs um CAR-T-Zellen, die zusätzlich ein oder mehrere transgene Zytokine enthalten, deren Synthese aber erst am/im Tumor nach Bindung an ein Tumorantigen beginnt. Erreicht wird dies durch sog. NFAT-Einheiten (nuclear factor of activated T-cells), die ihrerseits nach entsprechender Stimulation eine Zytokin-Genexpression in der T-Zelle induzieren/steigern und damit die sekundäre Immunantwort deutlich verstärken. Derzeit liegen die Zytokine IL-7, IL-12, IL-15 und IL-18 im Fokus der Erforschung. IL-12 verstärkt beispielsweise die Zytotoxizität von T- und NK-Zellen und induziert deren IFN-g Freisetzung. Die Zytokinfreisetzung kann auto- oder parakrin erfolgen. TRUCKs werden auch als CAR-T-Zellen der 4. Generation bezeichnet.
3.10.4 Wie kommen die Zellen in den Patienten?
Nach der erwähnten Leukapharese erfolgt die Produktion der CAR-T-Zellen in spezialisierten Zentren. Dort erfolgt auch eine Aufreinigung, Kultivierung, Stimulation und die Transduktion mit den Genfähren (CAR-Vektoren). Nach Fertigstellung erfolgen Kryokonservierung und qualitätssichernde Untersuchungen wie Freiheit von Bakterien, Endotoxinen, Mykoplasmen, Bestimmung der Zellzahl (die Dosierung beträgt X mal 106 bis 108 Zellen), Vektorkopienzahl pro Zelle und ein Funktionstest mittels Zytokinsekretions- oder -zytotoxizitäts-Assay. Der Transfusion geht eine dreitägige Konditionierung mittels nicht-myeloablativer, lymphodepletierender Chemotherapie, z.B. Cyclophospamid und Fludarabin (FC) voraus. Sie führt zu einer Reduzierung der Anzahl der körpereigenen Immunzellen und schafft im Körper des Patienten günstige Bedingungen für die Expansion der infundierten CAR-T-Zellen. Zwischen Konditionierung und Retransfusion wird noch ein bis zwei Tage pausiert, um die Proliferation und Funktionalität der CAR-T-Zellen möglichst nicht zu beeinträchtigen.
CAR-T-Zellen werden durch die Tumorzellbindung aktiviert und werden zu zytotoxischen T-Zellen. Bei Erkennung der Tumorzellen zerstören CAR-T-Zellen diese. Gleichzeitig beginnen sie aufgrund der Aktivierung zu proliferieren. Letzteres macht CAR-T-Zellen zu einem therapeutischen Wirkstoff, der sich im Patienten vermehrt und so über die Zeit zu einer „Wirkstofferhöhung“ im Patienten führt. Man geht nämlich davon aus, dass nur ein Bruchteil der infundierten Zellen aktiviert wird, dann proliferiert und für die therapeutische Wirkung sorgt. Nach Verschwinden der Tumorzellen können CAR-T-Zellen zudem länger im Patienten persistieren und – theoretisch – bei Wiederauftreten des Tumors erneut aktiv werden. Andererseits sind nach einer CAR-T-Zelltherapie durchaus Rezidive beobachtet worden, so dass sie nicht das High-Tech-Allheil-&-Wundermittel sind.
3.11 Vakzinierung
3.11.1 Prophylaktische Vakzinierung
Norbert Schleucher
Eine Art Menschheitstraum ist es, den Körper prophylaktisch gegen die Entstehung von Krebs zu schützen. Das wiederum setzt die genaue Kenntnis der Ursachen und der Mechanismen voraus. Ein tumorgener Mechanismus ist die Infektion mit bestimmten Viren. Wenn auch eine Tumorinduktion durch Viren insgesamt selten ist, so tragen Viren dennoch ursächlich zu mindestens 15 % der Tumorentstehung bei. Davon sind 70 % mit dem humanen Papillomavirus in Form von Zervixkarzinomen assoziiert. Von den zirka 100 bekannten HPV-Typen können mindestens 14 Gebärmutterhalskrebs auslösen, wobei HPV 16 + 18 für zirka 70 % aller Zervixkarzinome verantwortlich sind.
Derzeit versucht man im Rahmen von Prophylaxeprogrammen, diese Erkrankung durch Impfungen von jungen Mädchen auszurotten. Die Impfung wirkt optimal, wenn die jungen Frauen noch nicht mit HPV infiziert sind. Mittlerweile gibt es mit Cervarix® und Gardasil® zwei prophylaktische HPV-Impfstoffe mit gutem Sicherheits- und Wirksamkeitsprofil. Das Zervixkarzinom gehört weltweit zu den häufigsten HPV-assoziierten Karzinomen [5].
Die nachfolgende Graphik stellt den Anteil der durch HPV-Infektionen verursachten Krebserkrankungen weltweit dar [6, 7].
Die Inzidenz des Zervixkarzinoms liegt in Deutschland bei 5000–7000 Frauen/Jahr [8, 9].
Die Mortalität liegt bei 1700–2000 Frauen/Jahr [8, 9].
Zervixkarzinome sind zu fast 100 % HPV-assoziiert [6–9].
Die beiden zugelassenen HPV-Impfstoffe sind zur Anwendung ab einem Alter von 9 Jahren zugelassen. Beide Impfstoffe zeigen gute Wirksamkeit bei guter Sicherheitsdatenlage [8, 10–11].
3.11.2 Therapeutische Vakzinierung
Norbert Schleucher
Einen therapeutischen Ansatz stellt die sogenannte Impfung gegen Tumorzellen oder gegen wachstumsstimulierende Sekretionsprodukte dar, wie z. B. die Vakzinierung bei Tumoren des Gastrointestinaltraktes gegen wachstumsstimulierende Botenstoffe. Als Beispiel sei hier die Vakzinierung gegen Gastrin bei Magen- und Pankreaskarzinomen genannt. Bei diesen Tumoren ist der Gastrinrezeptor auf der Zelloberfläche überexprimiert und durch Bindung von Gastrin, welches im Serum zirkuliert, wird das Wachstum der Tumorzellen stimuliert. Therapeutisch wird hier ein an Diphtherietoxin gekoppeltes Gastrin (G17DT = Gastrimune) intramuskulär verabreicht. Dadurch werden bei etwa 85 % der Patienten neutralisierende Antikörper gegen die Aminosäurensequenz des Gastrinmoleküls gebildet. Somit wird das zirkulierende Gastrin auf immunogenem Wege reduziert, wodurch eine Wachstumsstimulation unterbleibt. Auch wird bei diesen Tumoren die Kombination der Vakzinierung mit etablierten Zytostatika geprüft.
Weitere Entitäten, bei denen Vakzinierungsansätze klinisch getestet werden, sind z. B. das Bronchialkarzinom, das Prostatakarzinom und das kolorektale Karzinom.
Angriffspunkte immuntherapeutischer Ansätze beim Bronchialkarzinom stellen MAGE-A3 (Melanoma-Antigen E A3), MUC-1 (mucinöses Glykoprotein) und das EGF-Protein dar. Das MAGE-A3-ASCI (MAGE-A3 antigen-specific cancer immunotherapeutic) richtet sich z. B. gegen das natürliche Antigen MAGE-A3, welches beim Melanom entdeckt wurde, aber auch auf verschiedenen anderen Tumoren, wie z. B. dem nicht-kleinzelligen Lungenkarzinom, exprimiert wird. Das Prinzip gleicht dem einer Impfung: Gentechnisch nachgebaute spezifische Tumorantigene werden zusammen mit einem Adjuvans injiziert, welches die Tumorantwort verstärkt. Dadurch wird die Bildung von spezifischen Antikörpern und T-Zellen angeregt, die die jeweiligen Tumorzellen erkennen und zerstören. Eine groß angelegte Phase-III-Studie (MAGRIT) prüft den Stellenwert dieses Konzepts momentan in der Adjuvanz beim NSCLC.
Beim Prostatakarzinom steht mit dem prostataspezifischen Antigen (PSA) ein hochspezifisches Ziel für die Vakzinierung zur Verfügung. Auch hier versuchen Phase-III-Studien einen Überlebensvorteil für vakzinierte Patienten mit hormonrefraktärem Prostatakarzinom zu zeigen.
Beim kolorektalen Karzinom befindet sich mit ALVAC-CEA eine Vakzinierung gegen das CEA-Oberflächenantigen in klinischer Erprobung. Dabei wird CEA und ein zusätzlich stimulierendes Molekül an ein Kanarienpockenvirus gekoppelt, sodass nach intradermaler Applikation eine Immunantwort gegen CEA ausgelöst wird. Auch bei diesem Ansatz wird eine adjuvante Vakzinierungsstrategie diskutiert.
3.12 Wirkprinzip Antiangiogenese (mit moAK und nmKI)
Jürgen Barth
Antiangiogenese: Ab einer gewissen Tumorgröße reicht die passive Diffusion für die Versorgung des Tumors mit Nährstoffen nicht mehr aus. Tumoren versuchen daher durch Gefäßneubildungen (Neoangiogenese) Anschluss an das Versorgungssystem des Wirtes zu bekommen. Dabei kommt es zuvor zur Sekretion von proangiogenen Zytokinen, wie beispielsweise den vaskulären endothelialen Wachstumsfaktoren (VEGF) mit diversen Subtypen. Für die neoplastische Gefäßbildung sind des Weiteren der PDGF- und FGF-Rezeptor von Bedeutung. Nach Anschluss an das Gefäßsystem ist der Tumor nicht nur gut mit Nährstoffen versorgt, er kann über diesen Weg auch metastasieren. Die Gefäßneubildung kann an unterschiedlichen Punkten gehemmt werden.
Mit dem moAK Bevacizumab wird im Blutkreislauf der zirkulierende VEGF abgefangen und erreicht seinen Rezeptor somit nicht. Gleiches wird mit Aflibercept erreicht. Es handelt sich um ein Fusionsprotein, bestehend aus den VEGF-bindenden Teilen der extrazellulären Domänen der humanen VEGF-Rezeptoren 1 und 2, fusioniert mit dem Fc-Teil des humanen IgG1. Aflibercept wird in der Literatur auch als VEGF-Trap – „Ligandenfalle“ – bezeichnet. Ramucirumab blockiert den VEGF-Rezeptor 2, sodass VEGF nicht mehr binden kann. Die nmKI, wie z. B. Lapatinib, hemmen dagegen, wie beschrieben, die intrazelluläre, proangiogene Signaltransduktion.
Lange bevor die ersten antiangiogenen Wirkstoffe entwickelt waren, glaubte man an einen eleganten, „smarten“, untoxischen Therapieansatz. Die Vorstellung des selektiven „Aushungerns“ der Tumoren ist jedoch falsch. Im Gegenteil, der Selektionsdruck steigt und verursacht bei einer „Übertherapie“ Resistenzen. Beobachtete Nebenwirkungen von Angiogenesehemmern umfassen (allgemein/Klasseneffekte):
- Bluthochdruck
- arterielle Thrombosen
- venöse Thrombosen
- Lungenembolien
- Wundheilungsstörungen
- Schwindel
- thromboembolische Ereignisse (z. B. Herzinfarkte)
- Blutungen
- Perforationen an Schwachstellen (nicht OP-bedingt; Peritonealkarzinose, Kolitis)
Das RPLS-Syndrom (RPLS = reversibles postanteriores Leukenzephalopathiesyndrom, zur Aussprechbarkeit auch PRES genannt), wahrscheinlich aufgrund von Bluthochdruck („Rindenblindheit“), wurde anfangs nur dem Bevacizumab zugeschrieben. Mittlerweile ist diese schwerwiegende Nebenwirkung auch für die mehr oder weniger stark antiangiogen wirkenden nmKI Axitinib, Bevacizumab, Regorafenib, Sorafenib, Sunitinib u.a.m. in der FI beschrieben (siehe hierzu auch Teil Nebenwirkungen Abschnitt Kardiotoxizität). Die Nebenwirkungen erklären sich auch dadurch, dass es derzeit keinen Wirkstoff gibt, der selektiv und alleinig die Gefäßneubildung des Tumors hemmt.
3.13 Zur Namensgebung von Wirkstoffen („-inib“, „-tinib“, „-anib“, „-mab“, „-mub“)
Wirkstoffnamen werden auch mit den Begriffen „InteRNAtionaler Freiname“ oder „INN (international nonproprietary name)“ oder „Generischer Name“ bezeichnet. Diese Bezeichnungen werden von der Weltgesundheitsorganisation (WHO) vergeben. Bei der Wahl des Namens hat der Entdecker zwar ein Vorschlagsrecht, es werden aber verschiedene Präfixe und Suffixe verwendet, um die Zugehörigkeit zu einer bestimmten Wirkstoffklasse anzuzeigen. Das geschieht entweder aus chemisch-struktureller Sicht oder aufgrund der Wirkungsweise.
Bei den smKI erkennt man an der Wortendung (Suffix) -tinib, dass es sich um einen Hemmstoff von Tyrosinkinasen handelt. Wirkt der smKI antiangiogen, so wird er mit dem Suffix -anib versehen, auch wenn die gehemmte Kinase eine Tyrosinkinase ist. An der Endung -tinib erkennt man also das pharmakodynamische Wirkziel, an der Endung -anib die Gesamtwirkweise (Antiangiogenese; siehe auch [12]). So hemmt Lapatinib die Tyrosinkinase-Domäne des HER2- und des EGF-Rezeptors. Pazopanib hemmt zwar auch Tyrosinkinasen (VEGFR-1, -2 und -3, c-kit sowie den PDGFR), wegen der antiangiogenen (Haupt-)Wirkung lautet das Suffix -anib. Ein derzeit „ungelöstes Problem“ stellt die Substanz Sorafenib dar, von der man der Meinung war, sie würde nur die unterschiedlichen Serin-/Threoninkinasen der RAF-Familie beeinflussen, was sich schon im Namen widerspiegelt. Nachträglich wurde jedoch auch eine Hemmwirkung auf unterschiedliche Tyrosinkinasen nachgewiesen.
Die moAK erkennt man an der Endung -mab für monoclonal antibody. Einer der ersten klinisch eingesetzten moAK war Muromonab-CD3 (Orthoclone OKT 3®) gegen akute Abstoßungsreaktionen von allogenen Nieren-, Herz- und Lebertransplantaten.
Nimmt man den INN-Namen auseinander, so steht:
- muro für Maus bzw. von der Maus (murin)
- monab für monoklonaler Antikörper (monoclonal antibody)
- mab allerdings auch für monoklonaler Antikörper (monoclonal antibody)
Nach ihrer Herkunft können die moAK am Namensstamm erkannt werden. So steht:
- -a- mab für einen AK von der Ratte
- -e- mab für einen AK vom Hamster
- -i- mab für einen AK von Primaten
- -o- mab für einen AK von der Maus (mouse), Beispiel: Oregovomab, Ibritumomab
- -u- mab für einen AK vom Menschen (voll human), dieser wird jedoch nicht vom Menschen gewonnen, sondern durch rekombinante Gentechnologie hergestellt, Beispiel: Ipilimumab, Ramucirumab; siehe daher unten unter Chimäre AK.
Chimäre AK (hergestellt aus genetisch verschiedenen Geweben oder Organismen) werden wie folgt benannt:
- -axo- mab für einen Hybridantikörper Ratte x Maus (rat x mouse) Beispiel: Catumaxomab (Zulassung zurückgezogen)
- -xi- mab (z. B. Rituximab, Basiliximab, Infliximab) für „irgendeine“ Chimäre, z. B. Mensch x Maus („ximeric“ monoclonal antibody)
- -zu- mab (z. B. Trastuzumab) für einen humanisierten, also mittels rekombinanter Gentechnologie hergestellten AK („xi-meric“, „zi-meric humanized“ monoclonal antibody)
- -xi- zumab für eine Kombination aus chimärem und humanisiertem AK (derzeit kein Beispiel verfügbar)
- -hu- mab oder -u- mab kennzeichnen voll humanisierte moAK (Abbildung rechts). Diese werden aus Mäusen mit „menschlichem Immunsystem“ gewonnen. Bei diesen modifizierten Mäusen wurden die mauseigenen Antikörpergene inaktiviert und durch die entsprechenden menschlichen Gene funktionell ersetzt. Diese transgenen Mäuse generieren vollständig menschliche Antikörper (XenoMouseTM). Daneben existiert noch das Trimera-Maus-System. Hierbei handelt es sich um normale Mäuse, die durch Ganzkörperbestrahlung immunoinkompetent gemacht wurden. Ein Beispiel für einen voll humanisierten AK ist das Panitumumab.
Aus einem INN-Namen eines AK können noch weitere Dinge herausgelesen werden. Mit der Vorsilbe (Präfix) ist ein Hersteller variabel (Aussprechbarkeit sollte noch gewährleistet sein). Im Mittelteil des Namens (Infix) steckt, im Unterschied zu den nmKI (siehe oben), das therapeutische Ziel, gefolgt von der Herkunft. Die Nomenklatur wurde im Laufe der Zeit modifiziert bzw. geändert. Die INN-Namen der moAK die zu den Zeitpunkten zugelassen waren, wurden jedoch nicht geändert. Eine Übersicht gibt die folgende Tabelle.
| Präfix | Therapeutisches Ziel / Unterstamm | Quelle / Unterstamm bis 2017 | Stamm | ||||
| Alt (~1993) | 2009 | Neu 2017 | Bedeutung | Bedeutung/Herkunft | |||
| Variabel | — | -ami- | -ami- | Serum Amyloid Protein (SAP) Amyloidosis (prä-Unterstamm) | -a- | Ratte | -mab -pab |
| -anibi- | — | — | Angiogenese (Inhibitor) | -e- | Hamster | ||
| -ba(c)- | -b(a)- | -ba- | Bakteriell (bacterial) | -i- | Primaten | ||
| -ci(r)- | -c(i)- | -ci- | Kardiovsakulär (cardiovascular) | -o- | Maus | ||
| -fung- | -f(u)- | fung- | Fungal | -u- | Human | ||
| -d(e)- | -de- | — | Endokrin | -xi- | chimär (chimeric; human + foreign) | ||
| -gr(o)- | -gros- | gros- | Skelettale und Muskelmassen verwandte Wachstumsfaktoren und Rezeptoren skeletal muscle mass related growth factors and receptors (prä-Substamm) | -zu- | humanisiert | ||
| -ki(n)- | -k(i)- | -ki- | Interleukin | xizu- | chimär/humanisiert hybrid | ||
| -les- | — | — | Inflammatorische Läsionen inflammatory lesions | axo- | Ratten-Maushybrid (s. Trifunktionaler Antikörper) | ||
| -li(m)- | -l(i)- | -li- | Immunmodulierend (immunomodulating) | ||||
| -mul- | — | — | Muskuloskelettales System (musculoskeletal system) | ||||
| ne(u)(r)- | -n(e)-* | -ne- | Neural | ||||
| -os- | -s(o)- | -os- | Knochen (ossär, bone) | ||||
| -co(l)- | -t(u)- | -ta- | Dickdarmtumore (colonic tumor) | ||||
| -go(t)- | Testikulärer Tumor (testicular tumor) | ||||||
| -go(v)- | Ovarialtumor (ovarian tumor) | ||||||
| -ma(r)- | Mammilärer Tumor (mammary tumor) | ||||||
| -me(l)- | Melanom (melanoma) | ||||||
| -pr(o)- | Prostatakarzionom (prostate tumor) | ||||||
| -tu(m)- | Verschiedene/sonstige Tumore (miscellaneous tumor) | ||||||
| -toxa- | tox(a)- | toxa- | Toxin (gekoppelter moAK) (toxin) | ||||
| — | — | -vet- | Veterinärmedizinische Verwendung veterinary use (prä-Stamm) | ||||
| -vi(r)- | -v(i)- | -vi- | viral | ||||
Diese Tabelle darf aber nicht einfach von links nach rechts gelesen werden, sondern man muss sich den gesamten AK-Namen zusammenstückeln. Beispiele basierend auf obiger Tabelle zeigt die Tabelle unterhalb.
| Präfix | Therapeutisches Ziel / Unterstamm | Quelle / Unterstamm bis 2017 | Stamm | |||
| Variabel | Alt (~1993) | 2009 | Neu 2017 | Bedeutung/Herkunft | ||
| Ce | -tu(m)- | t(u)- | -ta- | -xi- | chimär | –mab |
| Den | -os- | -s(o)- | -os- | -u- | human | –mab |
| Ipi | -li(m)- | -l(i)- | -li- | (m) -u- | human | -mab |
| Pembro | -li(m)- | -l(i)- | -li- | -zu- | humanisiert | –mab |
| Ramu | -ci(r)- | -c(i)- | -ci- | (r) -u- | human | –mab |
| Tafasi | -tu(m)- | -t(u)- | -ta- | humanisiert | –mab | |
| Tras | -tu(m)- | t(u)- | -ta- | -zu- | humanisiert | –mab |
| Belan | -tu(m)- | -t(u)- | -ta- | humanisiert | –mab (Mafodotin) | |
| Ca | tu(m)- | -t(u)- | -ta- | -axo- | Ratten-Maushybrid-AK (Trifunktionaler Antikörper) | –mab (Zulassung zurückgezogen) |
Das Subsystem, das der AK-Quelle vorangestellt ist, bezieht sich auf das therapeutische Ziel, wie Tumorart und/oder Organsysteme (kardiovaskulär, Skelett/Knochen). Der Term „Therapeutisches Ziel“ in obiger Tabelle impliziert jedoch nicht, welche Art der Pharmakodynamik der Antikörper ausübt. Es wird beispielsweise nicht unterschieden, ob es sich um einen therapeutischen, prophylaktischen oder diagnostischen AK handelt.
Im ursprünglichen System bestanden die Subterme meistens aus der Reihenfolge Konsonant-Vokal-Konsonant. Der letzte Buchstabe kann fallengelassen werden, wenn die Aussprechbarkeit Probleme mit sich bringt. Erkenntlich wird das durch einen in Klammern gesetzten letzten Buchstaben wie bei -ci(r)- für circulatory system, -li(m)- für immune system (lim steht dabei für lymphocyte) und -ne(r)- für nervous system. Dieser kann weggelassen werden. Das Weglassen kommt zum Tragen, wenn die darauffolgende Herkunftsquelle mit einem Konsonanten wie -zu- oder -xi- beginnt. Allerdings werden nicht alle Terme für die therapeutischen Subterme in ihrer verkürzten Form verwendet. So wird -mul- (musculoskeletal system) nicht zu -mu- reduziert, da kein chimärer oder humanisierter Antikörper einen INN-Namen bekommen hat (bisher). Kombinationen von Ziel- und Herkunftssubterm führen zu Endungen wie -limumab (immune system, human) oder -ciximab (circulatory system, chimeric, Konsonant doppeltes „m“ im ersten, „r“ im zweiten Fall weggelassen). Nach 2009 wurden kürzere Zielsubterme eingeführt. Sie bestehen meistens aus einer Konsonant-Vokal-Kombination, wobei der Vokal weggelassen wird, wenn der Herkunftssubterm mit einem Vokal beginnt. Humane AK, die das Immunsystem als therapeutisches Ziel haben, bekommen Namen die auf -lumab- enden anstatt auf -limumab- (Nivolumab statt Nivolilumab, 2015 zugelassen; vgl. Ipilimumab, 2011 zugelassen).
Merke:
Nach Modifikation des Nomenklatursystems sind zugelassene moAK hinsichtlich ihres INN Namens nicht umbenannt worden.
Um die Namen aussprechbar zu machen oder z. B. Namensähnlichkeit zu vermeiden, besteht die Möglichkeit, die Originalnamen silbenweise zu permutieren. Macht man das geordnet, entsteht ein „Lateinisches Quadrat“, wie in nachfolgender Tabelle am Beva-ci-zu-mab exemplarisch vorgestellt.
| BE | VA | CI | ZU | MAB |
| VA | CI | ZU | MAB | BE |
| CI | ZU | MAB | BE | VA |
| ZU | MAB | BE | VA | CI |
| MAB | BE | VA | CI | ZU |
3.14 Mechanismen der Zytostatikaresistenz
Die in der antitumoralen Chemotherapie eingesetzten Substanzen stammen aus unterschiedlichen Quellen. Einige sind rein synthetisch, bei anderen handelt es sich um Naturstoffe oder deren Derivate. Sie greifen in diverse biochemische Prozesse der Zelle mit unterschiedlichen Angriffspunkten ein (siehe auch Abschnitt Wirkungs- und Resistenzmechanismen). Die Substanzen scheinen eine Gemeinsamkeit zu haben: Krebszellen haben die Fähigkeit, gegen praktisch jede Substanz(klasse) resistent werden zu können (erworbene oder intrinsische Chemotherapieresistenz).
3.14.1 Kinetische Resistenz
Von kinetischer Resistenz spricht man, wenn viele Tumorzellen sich in der sog. G0-Phase des Zellzyklus befinden. Sie werden auch als „ruhende“ Zellen bezeichnet. Der Tumor an sich wächst langsam und ist nach der Diagnose nicht zwingend behandlungsbedürftig (Beispiel: indolente Non-Hodgkin-Lymphome → Haarzellenleukämie). Allerdings sind die Tumorzellen temporär unempfindlich gegen Antineoplastika, da die G0-Zellen prinzipiell jederzeit wieder zur Proliferationsfraktion rekrutiert werden können. Wenn das eintritt, wird der Tumor auch wieder „aktiver“, er wächst schneller. Diese kinetische Resistenz erklärt die relative Chemotherapieresistenz langsam wachsender, großer Tumore. Der Anteil der Tumore, die einen hohen Anteil an G0-Phasen-Zellen aufweisen, wird auch als q-Kompartiment bezeichnet (q = „quiescent“; ruhend, schlafend).
3.14.2 Genetische Resistenz
Die genetische Resistenz wird auch als primäre oder intrinsische Resistenz bezeichnet. Es besteht eine Unempfindlichkeit der Zellen bereits vor einer Zytostatikaexposition. Diese Unempfindlichkeit ist überwiegend genetisch bestimmt, die Ursachen dafür unklar. Diese genetische Resistenz ist nicht reversibel. Das Nierenzellkarzinom beispielsweise reagiert praktisch auf keins der klassischen, zytotoxischen Antitumortherapeutika.
3.14.3 Erworbene Resistenz (Acquired Resistance)
Die erworbene Resistenz ist der Empfindlichkeitsverlust von Zellen gegenüber einem oder mehreren Zytostatika nach oder während einer Exposition mit dem jeweiligen Agens. Im klinischen Sprachgebrauch wird überdies von Resistenz gesprochen, wenn ein Tumor auf eine Chemotherapie nicht mit einer Größenabnahme reagiert oder gar an Größe zunimmt (siehe auch Abschnitt Remissionsdefinitionen). Das bedeutet, dass das Nettotumorwachstum den Zellverlust übersteigt, nicht jedoch, dass alle Tumorzellen gegen die Zytostase resistent sind. Mit einer CTX können auch resistente Subklone eines Tumors selektiert werden, was schwer zu beurteilen ist. In der Konsequenz muss das Therapieregime geändert werden.
3.14.3.1 Voraussetzungen für eine zytotoxische Wirkung intrazellulär angreifender Moleküle
Das allgemeine Prinzip, damit ein Wirkstoff letale Wirkung auf eine Zelle hat, ist, dass
- der Wirkstoff durch die Zellmembran in die Zelle eindringt,
- dort teilweise einer Aktivierung bedarf und dann
- an das intrazelluläre Zielmolekül oder die Zielstruktur binden muss.
Man schätzt, dass zwischen 10.000 und 100.000 Moleküle eines Zytostatikums intrazellulär vorhanden sein müssen, um einen tödlichen Zellschaden zu verursachen.
Durch Modifikationen dieser allgemeinen Prinzipien kann die Arzneistoffwirkung vermindert werden. Hoch resistente Zelllinien bedienen sich einer Kombination der im Folgenden noch erläuterten Prozesse.
3.14.4 Verminderter Arzneistofftransport durch die Zellmembran
Obwohl einige Wirkstoffe durch passive Diffusion in die Zelle gelangen, sind viele andere Substanzen auf einen erleichterten oder gar aktiven Transport durch die Zellmembran angewiesen. Die sogenannten Antimetaboliten, die den natürlichen Substraten strukturell ja sehr ähnlich sind, werden beispielsweise über das gleiche Transportsystem, teilweise aktiv (d. h. unter Energieverbrauch), in die Zelle eingeschleust. Dieser Vorgang beinhaltet üblicherweise das Anbinden an ein (Rezeptor-)Protein auf der Zellmembran. Dieser Protein-Wirkstoff-Komplex durchwandert die Zellmembran, der Wirkstoff wird dann in das intrazelluläre Milieu freigesetzt. Durch Veränderungen in der Proteinstruktur des Bindeproteins oder durch Reduktion der Anzahl der Rezeptormoleküle auf der Zellmembran kommt es zu einer deutlichen Verminderung des aktiven Transports von Wirkstoffen in die Zelle. Daraus kann eine unzureichende Arzneistoffkonzentration in der Zelle resultieren – die erwünschte Zytotoxizität bleibt aus. Klinische Beispiele für diese Art der Resistenz betreffen Methotrexat und andere Antimetabolite.
3.14.4.1 Verminderter Arzneistofftransport durch veränderte Influxtransporter
Moleküle, die natürlichen Metaboliten nicht ähneln, gelangen ebenfalls in Körperzellen. Dies geschieht durch sog. Arzneistofftransporter, genauer Influxtransporter. Aus das kann aktiv erfolgen oder durch erleichterte Diffusion, also energieunabhängig entlang eines Konzentrationsgradienten. Man kennt über 400 Transportproteine, die in die „Superfamilien“ ABC- und SLC-Transporter eingeteilt werden.
3.14.4.2 ABC-Transporter
„ABC“ steht für „ATP-binding Cassette“, eine Proteindomäne, die ATP binden kann. Dieses wird dann für den Transport gespalten. ABC-Transporter arbeiten also primär aktiv. Es sind reine Effluxtransporter – sie transportieren Stoffe aus der Zelle heraus in den Extrazellulärraum (siehe Abschnitt Erhöhte intrazelluläre Entgiftung durch Effluxpumpen).
3.14.4.3 SLC-Transporter
„SLC“ steht für „Solute Carrier“. Auch SLC-Transporter vermitteln sekundär aktiv oder durch erleichterte Diffusion den Influx und Efflux von Arzneimitteln und somit den Transport durch polare Zellen. Die Influxfunktion überwiegt jedoch bei den SLC Transportern, wie beispielsweise die Resorption des Medikaments aus dem Darmlumen und anschließend die Sekretion ins Blut.
Merke:
Die ABC-Transporter verbrauchen ATP als Energiequelle und pumpen die Fremdstoffe aus der Zelle heraus (Efflux).
Die SLC-Transporter sorgen mittels Konzentrationsgradienten für die Aufnahme in die Zelle (Influx).
Imatinib wird beispielsweise durch den Influxtransporter OCT1 (Organischer Kationentransporter; organic cation transporter) in die Zelle transportiert. Durch eine verminderte Affinität zum Imatinibmolekül oder verminderter OCT1 Expression auf der Zellmembran im Rahmen der angenommenen Zytostatikaresistenz gelangt nicht mehr ausreichend Imatinib in die (CML-)Zelle. Anders ausgedrückt, die extrazelluläre Imatinibkonzentration müsste erhöht werden, was aber nicht mehr tolerabel wäre. Influxtransporter können auch durch Medikamente gehemmt werden. Wie in Zellkulturen nachgewiesen, hemmt der Alpharezeptorenblocker Prazosin OCT1 und somit die Imatinibaufnahme in die CML-Zellen. Diclofenac hingegen fördert die zelluläre Aufnahme.
3.14.5 Reduzierte intrazelluläre Stoffaktivierung
Diverse Zytostatika kommen als Vorstufen in die Zellen. Sie müssen intrazellulär aktiviert werden, um ihre Wirkung zu entfalten. Purin- und Pyrimidinanaloga müssen intrazellulär zum korrespondierenden Triphosphat aktiviert werden. Sie können nicht als solches appliziert werden, da die Nukleotide die Zellmembran nicht durchdringen und auch nicht per Transportsystem in die Zelle geschleust werden. Durch Veränderung der zur Phosphorylierung notwendigen Kinasen bzw. Phosphoribosyltransferasen hinsichtlich Umsatzrate und/oder Selektivität wird weniger Substanz aktiviert. Eine Erhöhung der Dosis ist sinnlos, da der kritische Schritt – die metabolische Aktivierung – reduziert ist. Als weiteres Beispiel einer Resistenz aufgrund verminderter Substanzaktivierung sei die zelluläre Unempfindlichkeit gegenüber Irinotecan (Synonym: CPT 11) durch reduzierte Aktivität des Enzyms Carboxylesterase genannt. Dadurch wird CPT 11 nicht in ausreichenden Mengen zum aktiven Metaboliten SN 38 verstoffwechselt. Nicht außergewöhnlich ist auch der vollständige Verlust eines oder mehrerer solcher Aktivierungsenzyme. Vom Standpunkt der Zelle aus sind solche Enzymdeletionen nicht letale Mutationen und führen zur Selektion hoch resistenter Zellklone.
3.14.6 Veränderte oder erhöhte Menge intrazellulärer Zielmoleküle
Viele Zytostatika erzeugen ihre Wirkung durch die Bindung an normale Zellenzyme, inaktivieren diese oder halten sie in einem physiologisch nicht funktionalen Zustand. Substanzen, die so wirken, binden stärker – mit höherer Affinität – an das Enzym, sodass das physiologische Substrat nicht umgesetzt werden kann. Beispiele hierfür sind Methotrexat (MTX) als Dihydrofolatreduktase-Hemmstoff oder Fluorouracil (5-FU) in Modulation mit Folinsäure zur Thymidylatsynthetase-Hemmung. MTX-resistente Zellen zeigen entweder erhöhte Spiegel an Dihydrofolatreduktase (intrazelluläre MTX-Menge reicht nicht aus) oder synthetisieren Dihydrofolatreduktase-Varianten, die durch MTX nicht (mehr) hemmbar sind. Um diese Resistenzen zu umgehen, müssten MTX-Dosen appliziert werden, die mit dem menschlichen Leben nicht mehr vereinbar wären.
Ein weiteres Beispiel für veränderte intrazelluläre Zielmoleküle ist das Genprodukt BCR-ABL bei der Philadelphia-Chromosom-positiven CML. Für diesen molekularen Treiber sind derzeit über 73 Punktmutationen beschrieben, sodass Imatinib vermindert (Dosiserhöhung ist notwendig) oder gar nicht mehr wirken kann. Diese Mutationen betreffen die ATP-Bindungstasche, die sogenannte Aktivierungsschleife oder das katalytische Zentrum. Beim Austausch der Aminosäure Threonin zu Isoleucin in Position 315 (T315I-Mutation) verlieren Imatinib, Nilotinib und auch der duale SCR-ABL-Inhibitor Dasatinib vollständig ihre Wirksamkeit. Durch eine Amplifikation des BCR-ABL-Gens wird die Wirkung der nmKI praktisch „übersteuert“ („zu viel“ BCR-ABL; siehe MTX).
3.14.7 Veränderte Menge extrazellulärer Zielmoleküle
Von dieser Art Resistenzmechanismus sind primär moAK betroffen (siehe Abschnitt Resistenzen gegen monoklonale Antikörper – non-Checkpoint-Inhibitoren). Das von der Leber synthetisierte Akute-Phase-Protein a-saures Glykoprotein (AGP) bindet Imatinib mit hoher Affinität. Dies führt zur Verringerung der freien Imatinibkonzentration, da Imatinib nur in freier Form aktiv von der Leukämiezelle aufgenommen wird. Wird im Rahmen einer CML vermehrt saures α-1-Glykoprotein gebildet, führt das zu verminderten Imatinib-Plasmaspiegeln, weil der Wirkstoff dadurch im peripheren Blut sozusagen „abgefangen“ wird. Die Bedeutung des APG als Resistenzmechanismus und Prognosefaktor für die Behandlung von Patienten mit CML in Blastenkrise und Bcr-Abl positiver ALL konnte mittlerweile sowohl in vitro als auch in vivo belegt werden.
3.14.8 Erhöhte intrazelluläre Entgiftung
Während bei der Arzneistoffaktivierung mit sogenannten Phase-I-Reaktionen die Wirkstoffe oxidiert (größte Bedeutung: Zytochrom P 450; Monooxygenasen), reduziert oder hydroxyliert und diese damit im Allgemeinen hydrophiler werden, handelt es sich bei Entgiftungsreaktionen vielfach um sogenannte Phase-II-Reaktionen. Hierbei kommt es zu Konjugationsreaktionen unter Beteiligung spezifischer Enzyme – den Transferasen. Wichtigste Konjugationsreaktionen sind die von Zytostatika mit Glutathion. Diese werden über Glutathion-S-Transferasen (GST) vermittelt. Hiervon gibt es beim Menschen eine ganze Reihe von Isoenzymen mit unterschiedlicher Spezifität. Es können z. B. eine erhöhte Substanzausscheidung und geringere antitumorale Aktivität resultieren, aber auch verstärkte Toxizität. Radikalbildende Zytostatika wie die Anthrazykline werden durch das GST-Redoxsystem, das physiologischerweise schädliche Peroxide und aggressive Radikale abfängt, in ihrer Effektivität gemindert.
Obwohl der Stellenwert der Glutathionsysteme im Resistenzgeschehen kontrovers diskutiert wird, kann man davon ausgehen, dass sie ihren Teil dazu beitragen. So dürften sie zytotoxische Effekte einiger Substanzen reduzieren und damit zumindest Bestandteil eines komplexeren Resistenznetzwerks sein.
3.14.8.1 Erhöhte intrazelluläre Entgiftung durch Effluxpumpen
Substrate können nicht nur aktiv in Zellen hinein transportiert werden, sondern auch aus ihnen heraus. Bei Arzneimitteln geschieht das über sog. „Drug Transporter“. Die bedeutendste Gruppe der „Uptake Transporter“ sind die Mitglieder der organischen Anionentransporter-Polypeptide (OATP) und die organischen Kationentransporter (OCT) (siehe Abschnitt SLC-Transporter). Effluxtransporter gehören zur Familie der ATP-binding-Cassette(ABC)-Transporter. Hierzu gehören das Breast Cancer Resistance Protein (BCRP) und das Multidrug Resistance Protein 2 (MRP2). Effluxtransporter für kationische Komponenten sind das Multidrug and Toxin Extrusion Protein 1 (MATE1) und 2-K. Mit hoher Wahrscheinlichkeit handelt es sich um natürliche Schutzmechanismen gegen Vergiftungen. Tumoren können eine erhöhte Menge an Effluxtransportern aufweisen. Sie können auch im Laufe der Chemotherapie induziert werden. Am besten untersucht ist in diesem Zusammenhang die Multidrug-Resistenz.
3.14.8.2 Der „Multidrug Resistance“-Phänotyp
Liegt eine Kreuzresistenz gegen verschiedene, chemisch nicht miteinander verwandte Substanzen vor, spricht man von „Multidrug-Resistenz“, kurz MDR (Mehrfachresistenz gegen unterschiedliche Arzneistoffe). Dabei unterscheidet man die klassische und die atypische MDR.
Klassische MDR: Bei der klassischen MDR findet sich in den Tumorzellen eine Überexpression eines membranären Zelloberflächenglykoproteins mit einem Molekulargewicht von 170 Kilodalton. Daher der Name P-Glykoprotein 170, kurz Pgp170, wobei P für Permeabilität steht. Auch dieses Protein gehört zur sogenannten Familie ATP-bindender Enzyme (ABC-Transporter). Sowohl Pgp170 als auch weitere Vertreter der Familie sind in der Natur weit verbreitet. Man findet sie bei Säugern, Malariaerregern und auch bei Hefen. Die Funktion von Pgp170 ist die einer „Ausflusspumpe“ im Sinne eines Entgiftungsmechanismus. Das für Pgp170 kodierende Gen (MDR1) ist in vielen Normalgeweben vorhanden, was die These eines physiologischen Entgiftungssystems unterstützt. Gewebe mit dem MDR1-Gen sind in folgender Tabelle wiedergegeben.
| Hohe Expression | Mittlere Expression | Niedrige bis gar keine Expression |
| Nebennierenrinde Niere (proximal) Leber (Gallengänge) Plazenta Kolon Dünndarm Gehirn (Endothelzellen) Hoden (Endothelzellen) Pankreas (Epithelzellen) Makrophagen | Nebennierenmark Trachea Lunge (Hauptbronchien) Prostata CD34+ Progenitorzellen Lymphozyten (CD4+, CD8+, CD56+) fetales Gewebe | Haut Skelettmuskulatur Herz Ösophagus Magen Ovarien |
Physiologische Substrate für Pgp170 sind beispielsweise Steroidhormone, Aflatoxin B1 und Bilirubin. Das Glykoprotein kann auch von Substanzen, die vom MDR-Phänotyp eigentlich gar nicht erfasst werden, wie z. B. Arsen oder Tetrachlorkohlenstoff (Vergiftungen), induziert werden. Zytostatika, die von der klassischen MDR erfasst werden, sind Naturstoffe bzw. deren Derivate wie:
- Anthrazykline und verwandte Verbindungen (Dauno-, Doxo-, Epi-, Idarubicin(ol), Amsacrin, Actinomycin D)
- Vinca-Alkaloide
- Epipodophyllotoxin-Derivate
- Kinase-Inhibitoren (z. B. Imatinib)
Da diese Art der Resistenz, wie oben erwähnt, auch induzierbar ist (beispielsweise durch subtherapeutische Zytostatikadosen), muss immer auf eine adäquate Dosierung geachtet werden. Auf Grund dieser Zusammenhänge resultiert der Satz: „Eine unterdosierte Chemotherapie ist meist schlechter als gar keine“.
Während eine P-gp Hemmung zeitlich limitiert ist, scheint eine Induktion bei einem Tumor(!) von allein, also durch Weglassen des Induktors, nicht reversibel zu sein. Möglicherweise ist das für den Tumor ein Selektionsvorteil im Darwin‘schen Sinne. Beispiel: Venetoclax erhöht durch P-gp Hemmung im Gastrointestinaltrakt die Bioverfügbarkeit und damit die Spiegel von Digoxin in für den Patienten kritischer Größenordnung. Es reicht aber ein Einnahmeabstand von 6 Stunden aus (Digoxin 6 Stunden vor Venetoclax), um diese Hemmwirkung aufzuheben [FPI Venetoclax].
Merke:
Eine P-gp-Induktion im Tumorgewebe bildet sich nach aktuellem Kenntnisstand nicht von alleine zurück. Auch dann nicht, wenn der Induktor weggelassen wird.
Vielfach sind P-gp Induktoren auch starke Induktoren für CYP3A4. So induzieren
- Carbamazepin
- Phenobarbital
- Phenytoin
- Rifampin
- Johanniskraut
neben CYP3A4 auch P-gp170.
Merke:
Johanniskraut ist ein starker CYP3A4 und P-gp 170-Induktor. Tumorpatienten sollten jegliche Johanniskrautprodukte, seien es Arzneimittel oder Nahrungsergänzungsmittel, strikt meiden.
Substrate verschiedener Arzneistofftransporter mit Augenmerk auf P-gp170 aus der Gruppe der modernen Kinase-Inhibitoren und Enzymhemmstoffe zeigt die nachfolgende Tabelle. Sie soll auch die Wichtigkeit des „Johanniskrautverbots“ für Tumorpatienten unterstreichen.
| INN | Drug-Transportersubstrat |
| Abemaciclib | P-gp, BCRP |
| Acalabrutinib | P-gp, BCRP |
| Afatinib | BCRP, P-gp |
| Alectinib | M4-Metabolit: P-gp |
| Asciminib | BCRP, P-gp, UGT2B7, UGT2B17 |
| Binimetinib | P-gp, BCRP |
| Bosutinib | P-gp |
| Brigatinib | P-gp, BCRP |
| Capmatinib | P-gp |
| Ceritinib | P-gp |
| Cobimetinib | P-gp, UGT2B7 |
| Dabrafrenib | BCRP, P-gp |
| Dacomitinib | P-gp, BCRP |
| Duvelisib | P-gp, BCRP |
| Encorafenib | P-gp |
| Entrectinib | M5-Metabolit: P-gp, BRCP |
| Erdafitinib | P-gp |
| Erlotinib | P-gp |
| Everolimus | P-gp |
| Fedratinib | P-gp |
| Futibatinib | P-gp, BCRP |
| Gefitinib | P-gp |
| Gilteritinib | P-gp |
| Glasdegib | P-gp |
| Ivosidenib | P-gp |
| Ixazomib | P-gp (low-affinity) |
| Lapatinib | BCRP, OATP1B1, P-gp |
| Larotrectinib | P-gp, BCRP |
| Lenvatinib | P-gp, BCRP |
| Momelotinib | P-gp, BCRP, OATP1B1/1B3 |
| Nilotinib | P-gp |
| Nintedanib | UGT1A1, UGT1A7, UGT1A8, UGT1A10, P-gp |
| Niraparib | P-gp, BRCP |
| Nirogacestat | P-gp |
| Olaparib | P-gp |
| Osimertinib | P-gp, BCRP |
| Pazopanib | BCRP, P-gp |
| Pemigatinib | P-gp, BCRP |
| Pralsetinib | P-gp, BCRP |
| Quizartinib | P-gp | AC886 BCRP |
| Reprotrectinib | P-gp |
| Ripretinib | DP-5439 Metabolit: P-gp, BCRP |
| Rucaparib | P-gp, BRCP |
| Selpercatinib | P-gp, BCRP |
| Selumetinib | BCRP, P-gp |
| Talazoparib | P-gp, BRCP |
| Tazemetostat | P-gp |
| Tepotinib | P-gp |
| Trametinib | (BSEP), P-gp |
| Tucatinib | P-gp, BCRP |
| Vemurafenib | P-gp, UGT |
| Venetoclax | P-gp, BCRP |
| Zanubrutinib | P-gp |
| BCRP = Breast Cancer Resistance Protein BSEP = Bile Salt Export Pump P-gp = P-Glykoprotein 170 UGT = UDP Glucuronosyltransferasen | |
Eine P-gp Induktion auf medikamentösem Wege zu blockieren oder rückgängig zu machen, ist bisher gescheitert. Es ließen sich lediglich diverse Substanzen identifizieren, mit denen das auf Zellkulturniveau gelang. Ein wesentlicher Grund ist, dass im Menschen Dosierungen benötigt werden, die nicht tolerabel sind.
Atypische MDR: Während sich die klassische MDR auf das Pgp170 bezieht, spricht man von atypischer MDR im Zusammenhang mit einem Glykoprotein von 190 Kilodalton Molekulargewicht, auch als MRP (Multidrug Resistance Protein) bezeichnet. Die atypische MDR bezieht sich des Weiteren auf alle zellulären Phänotypen, die irgendeine Art der pleiotropen Resistenz aufweisen, und ist überdies mit Mutationen, beispielsweise der Topoisomerase II, vergesellschaftet (Veränderungen in der Enzymaktivität, reduzierte Enzymspiegel; siehe oben). Vom MRP sind mittlerweile fünf weitere Isoformen identifiziert (MRP2–6). Von diesen Isoformen verfügen mindestens MRP2 und MRP3 über Effluxkapazität gegenüber Zytostatika (MRP2: Epipodophyllotoxine, Doxorubicin, Cisplatin; MRP3: Epipodophyllotoxine).
| Multidrug-Resistance-Protein-Subtyp | ||
| MRP1 | MRP2 | MRP3 |
| basolateralea Plasmamembran-oberfläche polarisierter Zellen | apikaleb Membranoberfläche von Gallengangskanälen, renales tubuläres Epithel | basolaterale Oberfläche diverser Gewebe, wie z. B. Kolon, Leber, Pankreas |
| a lateral(is): seitlich, von der Mitte(llinie) abgewandt b apikal: an der Spitze (Apex) eines Organs | ||
Sowohl die Rolle von MRP2 und MRP3 als auch die der derzeit weniger gut charakterisierten Isoformen MRP4–6 hinsichtlich ihrer klinischen Relevanz sind derzeit spekulativer Natur.
3.14.9 Erhöhte Reparaturkapazität
Zytostatika bedingen ihre Wirkung auch durch direkte Schäden an der DNS bzw. durch eine Beschädigung der strukturellen Integrität der DNS. Tumorzellen können hoch effektive Mechanismen zur Reparatur geschädigter DNS-Segmente entwickeln. Zelllinien, die Resistenz gegen Alkylanzien und Interkalatoren (z. B. Anthrazykline und verwandte Verbindungen) zeigen, verfügen meistens über eine erhöhte Reparaturkapazität gegen DNS-Schäden. Diese Mechanismen umfassen beispielsweise das Herausschneiden des geschädigten DNS-Segments (Exzision) und Ersatz durch die neu synthetisierte Basensequenz. Eine Resistenz gegenüber Cisplatin-DNS-Addukten konnte in bestimmten Fällen auf eine erhöhte Aktivität zweier DNS-Exzisionsreparaturenzyme zurückgeführt werden. Eine weitere wichtige Determinante der Alkylanzienresistenz ist die Fähigkeit der Zellen, Alkylierungen in bestimmter Position des Guanins spezifisch zu erkennen und gezielt zu entfernen. Von diesem Reparaturenzym sind in erster Linie die Nitrosoharnstoffverbindungen (ACNU, BCNU, CCNU) betroffen. Es können aber auch durch andere Zytostatika übertragene Methylreste und kurzkettige Alkylreste entfernt werden.
3.14.10 Unterdrückung apoptotischer Signalwege
Dass die Zytotoxiziät der verschiedenen Substanzklassen auf der Interaktion mit diversen molekularen Zielstrukturen basiert, darauf wurde mehrfach hingewiesen. Beispielhaft seien hier noch einmal genannt:
- Topoisomerase-II-Hemmung durch Epipodophyllotoxin-Derivate
- Addukte von Alkylanzien mit nukleophilen Zentren der DNS und von Proteinen
- Dihydrofolatreduktase-Hemmung mit der Folge reduzierter Purin- und Pyrimidinsynthese durch Methotrexat
Von diesen „Upstream“-Zielstrukturen abgesehen führen die meisten, wenn nicht gar alle Zytostatika durch eine „Downstream“-Signaltransduktion zum programmierten Zelltod (Apoptose). Die Apoptose ist ein energieverbrauchender, streng geregelter und gesteuerter Vorgang, der zum nicht nekrotischen Zelluntergang führt. Die Apoptose wird von proapoptotischen und antiapoptotischen Elementen – vornehmlich Proteinen – geregelt. Das p53-Genprodukt zählt zu den proapoptotischen Faktoren, ihm wird eine Schutzfunktion zugewiesen und es heißt auch „Wächter des Genoms“.
Zellen mit funktionellem Verlust von p53 zeigen reduzierte Apoptosefähigkeit und somit eine relative Resistenz gegenüber DNS-Schäden. Weitere wichtige Regulatoren der Apoptose sind Proteine der Bcl-2-Familie und PTEN (siehe oben). Obwohl die Vorgänge der Apoptose als solche derzeit noch nicht vollständig verstanden sind, können die Proteine, respektive die Genprodukte Bcl-2, Bcl-XL, Bcl-w, A1 und Mcl-1 der antiapoptotischen Reihe zugeordnet werden, während die Genprodukte Bax, Bal, Bad, Bik und Bid proapoptotisch wirken (siehe auch Abschnitt Hemmstoffe von Regulatorproteinen). Für erhöhtes Vorkommen von Bcl-2 und seinen Homologen wurde eine verstärkte Resistenz lymphoider Zellen gegenüber Kortikosteroiden, Bestrahlung und durch Arzneimittel verursachte DNS-Schäden gezeigt. Erhöhte Mengen von Bcl-2 und Bcl-XL reduzieren die Sensitivität gegenüber DNS-schädigenden Arzneistoffen. Ein Resistenzphänotyp, der durch erhöhtes Zellüberleben, verstärkte Toleranz gegenüber DNS-Schäden und genomischer Stabilität gekennzeichnet ist (siehe Abschnitt Einteilung der antineoplastischen Substanzen).
3.15 Resistenzen gegen monoklonale Antikörper – non-Checkpoint-Inhibitoren
Monoklonale Antikörper benötigen ein extrazelluläres Tumorantigen zum Andocken. Fehlt dieses oder ist es auf Grund von Mutationen derart verändert, dass der Antikörper nicht mehr daran binden kann, sieht man klinisch keine Wirksamkeit mehr und höchstwahrscheinlich eine verstärkte Systemtoxizität. Im Fall von Trastuzumab (Anti-HER2-Antikörper) kann eine Resistenz durch die Expression eines mutierten ErbB2-Rezeptors (p95HER2) vermittelt sein. Weil bei diesem trunkierten Rezeptor die extrazelluläre Domäne fehlt, kann der Antikörper nicht an den Rezeptor binden und es kommt zu keiner antikörpervermittelten Wachstumshemmung mehr. Bei einer weiteren Rezeptorvariante, der EGFRvIII, ist dieser konstitutiv aktiviert, auch ohne dass ein agonistischer Ligand an ihn binden muss. Durch die Synthese von Muc4, einem membranassoziierten Glykoprotein, kommt es zu einer sterischen Behinderung der Bindung von Trastuzumab an HER2, da Muc4 selbst an den Rezeptor bindet und seine Phosphorylierung bewirkt.
Das Tumorsuppressorgen PTEN hemmt den antiapoptotischen Signalweg. Das bedeutet, dass bei Ausfall (durch Mutation) von pten diese „Bremse“ nicht mehr funktioniert und das tumorale Überleben gefördert wird. Bei 50 % der HER2-überexprimierenden Tumoren kommen pten-Mutationen vor, die die tumorunterdrückende Wirkung ausschalten. Während der Antikörper Trastuzumab seine Wirksamkeit beim sogenannten pten-loss verliert, zeigt der nmKI Lapatinib unter diesen Bedingungen weiter Wirksamkeit.
Das 1981 erstmals beschriebene Onkogen KRAS aktiviert im EGFR-Signaltransduktionsweg nachgeschaltete Proteine, die das Wachstum, die Proliferation, die Differenzierung, die Apoptose der Zellen und die Angiogenese beeinflussen. Ist KRAS und/oder NRAS mutiert, wandelt es sich zu einem konstitutiv aktiven Protein, das eine Dauerstimulation des intrazellulären Signalweges bewirkt. Die Anti-EGFR-Antikörper Cetuximab und Panitumumab verlieren dann ihre Wirkung, weil die intrazelluläre Reaktionskaskade auch ohne externe Stimulation abläuft bzw. durch einen extrazellulär angreifenden Antikörper nicht mehr unterbrochen werden kann. Bei kolorektalen Karzinomen treten in zirka 35 % der Fälle solche Mutationen auf. Die Antikörper dürfen erst nach einer Testung auf „Wildtyp“ Pan-RAS eingesetzt werden.
Eine Mutation in der extrazellulären Domäne des EGF-Rezeptors (S492R) induziert eine Cetuximab-Resistenz, da sie mit der Bindung des Antikörpers an den Rezeptor interferiert. Diese Mutation findet sich nicht bei KRAS-Wildtyp-Tumoren, die noch nicht mit einem Anti-EGFR-Antikörper, speziell Cetuximab, behandelt wurden. Man geht daher von einer erworbenen (acquired) Resistenz aus. Daher macht es auch keinen Sinn, therapienaive Patienten mit KRAS-Wildtyp auf diese Mutation routinemäßig zu testen. Nach aktuellem Kenntnisstand besteht keine Kreuzresistenz zu Panitumumab.
3.16 Resistenzen gegen monoklonale Antikörper – speziell Checkpoint-Inhibitoren
Immuncheckpoint-Inhibitoren haben die Krebsbehandlung revolutioniert. Sie sind nicht nur jenseits der traditionellen, zytotoxischen Chemotherapie, sondern auch jenseits der „Targeted Therapies“ (siehe Abschnitt Targeted Therapies) anzusiedeln. Während bei einigen Patienten ein dramatisches Ansprechen zu verzeichnen ist, musste aber auch ernüchternd festgestellt werden, dass einige Tumore von Anfang an resistent sein können, oder, nach initialem Ansprechen, resistent werden können. Wie kommt das? Wir erinnern uns daran, dass die ICI – unkonjugierte Antikörper – nicht an ein Tumorantigen binden, sondern das Immunsystem, speziell T-Zellen aktivieren.
Die Resistenzen können genau wie bei den anderen Substazklassen in primäre – intrinsische – und sekundäre, oder erworbene Resistenz eingeteilt werden. Bei primärer Resistenz hat der Tumor niemals auf eine Checkpoint-Inhibitortherapie angesprochen.
Intrinsische und extrinsische Resistenz gegen ICI
Von intrinsischer Resistenz spricht man, wenn Tumorzellen relevante Prozesse der Immunerkennung, Signalpfade, Genexpressionen und ihre Antwort auf DNS-Schäden (DNA Damage Responses) verändern. Extrinsische Resistenz tritt außerhalb von Tumorzellen auf. So z. B. durch immunologische und nicht-immunologische Interaktionen zwischen T-Zellen, Makrophagen, MDSC, Immuncheckpoints, Enzymen, der Angiogenese und dem Mikrobiom.
3.16.1 Mechanismen intrinsischer Resistenz
3.16.1.1 Mangel an Neoantigenen
Tumorantigenspezifische T-Zellen sind im Tumorgewebe anwesend, ohne dass sie eine antitumorale Immunantwort zeigen. Das wird öfter bei Tumoren beobachtet, die nicht über ein erhöhtes TMB als auch nicht über Neoantigene verfügen, die einen fokussierten CD8+ T-Zellangriff stimulieren sollten. Genetische oder epigenetische Veränderungen seitens der Tumorzelle können nach initialem Ansprechen zur erworbenen Resistenz führen, da beispielsweise keine Tumorneoantigene mehr ausgebildet werden (Immune Escape). Von Hause aus niedrig immunogen und oftmals mit niedrigem TMB sind das Prostata- und Pankreaskarzinom. Hoch immunogen mit oftmals hohem TMB sind das Melanom und das nicht-kleinzellige Bronchialkarzinom. Das Nierenzellkarzinom ist dahingehend eine Ausnahme, dass es zwar hoch immunogen ist, jedoch überwiegend eine relativ niedrige TMB-Last mit sich trägt. Es müssen also weitere Faktoren am oftmals guten Ansprechen auf ICIs vorliegen.
Intrazelluläre Antigene werden über den MHC auf der Zelloberfläche präsentiert (siehe Abschnitt Zytotoxische T-Zellen). Eine Unterbrechung oder ein Zusammenbruch dieser Antigenpräsentation resultiert in einer Resistenz gegen T-Zell-mediierte Zytotoxizität. Dieser Antigenpräsentationsmechanismus kann beispielsweise durch Mutationen im Transporter für Antigenpräsentation (TAP) oder der MHC Struktur begründet sein. Des Weiteren sind Veränderungen in der Antigenpräsentationsmaschinerie und im b2M als auch eine Downregulation von MHC I beschrieben. Alterationen oder Mutationen, inkl. Loss-of-Function, im b2M führen beispielsweise zur Instabilität von MHC.
3.16.1.2 Epigenetische Veränderungen
Epigenetische Modifikationen wie Methylierung, Histonmodifikationen und das Ruhigstellen von Genen können die Expression immunrelevanter Gene verändern oder ausschalten. Dies führt zu Veränderungen in der Antigenprozessierung und -präsentation und damit zur Immunevasion und zur T-Zellerschöpfung (Exhaustion).
3.16.1.3 Veränderungen von Signalwegen
Die Aktivierung verschiedener onkogener Zellsignalwege kann das lokale, immunologische Antitumoransprechen reduzieren oder ganz verhindern. Der RAS/RAF/Mitogen aktivierte Proteinkinase (MAPK) Signalweg übermittelt Signale aus dem Extrazellulärraum in den Zellkern. Dort werden diese Signale in Zellproliferation, -teilung und -differenzierung übersetzt. Eine Dysregulation dieses Signalwegs ist mit maligner Transformation verbunden. Der MAPK-Signalweg hat überdies einen inhibitorischen Einfluß auf die T-Zellrekrutierung und -funktion, sodass es zu einer T-Zell-armen Tumorumgebung kommt. Aktivierung des MAPK-Signalwegs, beispielsweise durch EGFR- und KRAS-Mutationen oder EML4-ALK-Fusionen, kann zu einer Heraufregelung von PD-L1 auf den Tumorzellen führen, so dass es nahe liegt, dass der MAPK-Signalweg eine immunsuppressive Umgebung generiert und damit zur Resistenz gegenüber einer Immuntherapie führt.
Auch der PI3K/AKT Signalweg reguliert die Zellproliferation, oftmals mit der Information „stirb nicht“ und ist bei Tumoren abnormal aktiviert. Ein PTEN-Verlust verstärkt die PI3K-Signalgebung, was zur Produktion immunsuppressiver Zytokine führt. Ein solches Zytokin ist der proangiogene VEGF. VEGF führt zu verminderter T-Zellinfiltration in das TME. Tumorangiogenese ist also als immunsuppressiv anzusehen. Daher macht die Kombination aus Antiangiogenese und ICI Sinn, wie es mit der Zulassung von Pembrolizumab (anti-PD-1) in Kombination mit Axitinib gegen das Nierenzellkarzinom seit 2019 umgesetzt worden ist. Kurze Zeit später erfolgte die Zulassung des PD-L1-Antikörpers Avelumab, ebenfalls in Kombination mit Axitinib. Das Wirkprinzip ICI plus Antiangiogenese wird sicherlich weiterverfolgt werden.
Mutationen im PTEN-Signalweg sind weitere Mediatoren der Resistenz gegenüber einer ICI-Therapie. Hier ist der Wnt-Signalweg zu nennen, der essenzielle zelluläre Prozesse wie Proliferation und Migration mitsteuert. Abberationen in diesem Signalweg wurden als bedeutender Mechanismus der Onkogenese identifiziert.
Die Tumor-intrinsische Aktivierung des b-Catenin Signalwegs verursacht eine T-Zellenausgrenzung beim Melanom und damit eine Resistenz gegen eine ICI-Therapie.
IFN-g ist eines der T-Zelleffektorzytokinen. Es unterstützt die Inhibition des Tumorzellwachstums durch Unterdrückung der Zellproliferation, erleichterte Apoptose und Heraufregelung der Tumorantigenpräsentation via b2M. Letzteres wird über den JAK-STAT-Signalweg vermittelt. Eine andauernde IFN-g Stimulation überführt jedoch sensible Melanomzellen in einen resistenten Zustand. Eine Analyse der Tumorzellen von Patienten, die nicht auf eine Anti-CTLA4-Therapie ansprachen, zeigte eine erhöhte Rate an Mutationen in IFN-g Signalweggenen. Zu nennen sind der IFN-g-Rezeptor 1 und -2, JAK2 und der Interferon-regulatorische Faktor 1. Darüber hinaus führen JAK1/2-Mutationen zur primären Resistenz gegenüber einer PD-1-/PD-L1-Blockade durch den Verlust der IFN-g induzierten Expression von MHC I und PD-L1, sowie der Phosphorylierung des STAT-Transkriptionsfaktors. Tumorzellen können also die Effekte von IFN-g umgehen, indem Moleküle des IFN-g-Signalwegs heruntergeregelt oder durch Mutationen funktionsunfähig gemacht werden.
3.16.2 Mechanismen extrinsischer Resistenz
Extrinsische Faktoren, die zur Ausbildung einer Resistenz gegen Immuntherapeutika beitragen, stehen überwiegend mit dem TME in Verbindung. Weitere, inhibitorische Checkpoints, Zytokine, angiogene Faktoren, als auch vom Tumor abgelegene Faktoren wie das Mikrobiom des Patienten können zur extrinsischen Resistenz beitragen.
3.16.2.1 Abwesenheit von T-Zellen und der STING-Signalpfad
Dass intrinsische Faktoren die T-Zell-Infiltration in das TME reduzieren können, wurde im vorigen Abschitt beschrieben. Aber auch dendritische Zellen (DCs) und adere APCs spielen eine bedeutende Rolle bei der T-Zell-Infiltration ins TME. Der Stimulator of Interferon Genes (STING)-Signalweg trägt wesentlich zur angeborenen Immunerfassung (Innate Immune Sensing) von immunogenen Tumoren bei. Eine Heraufregelung des STING-Signalpfades resultiert in einer Aktivierung von APCs und in einem Priming von CD8+ T-Zellen gegen Tumore. Kommt es zu einer STING-Defizienz, wie im Mausmodell nachgewiesen, wird keine ausreichende T-Zellantwort gegen Tumore mehr ausgelöst. Grund ist eine insuffiziente Rekrutierung und Aktivierung von DCs. Dazu kommt, dass eine Inhibition von CTLA4 und PD-1/PD-L1 im STING-defizienten Mausmodell nicht mehr wirksam ist. Dem STING-Signalweg ist IFN-b nachgeschaltet. IFN-b ist essenziell für die Präsentation von Tumorantigenen gegenüber CD8+ Zellen.
3.16.2.2 Exosomaler PD-L1
Normalerweisebefindet sich der PD-1-Ligand auf der Oberfläche von Tumorzellen (siehe Abschnitt Programmed Cell Death Protein 1) und unterdrückt die Immunantwort im TME. PD-L1 wurde aber auch auf der Oberfläche extrazellulärer Vesikel (EV) gefunden, die von den Tumorzellen generiert worden sind. Sie wirken sozusagen als Anti-PD-L1-Antikörper, da sie diese abfangen und der Antikörper gar nicht mehr mit der Tumorzelle interagieren kann. Werden solche EV im peripheren Blut gefunden, ist möglicherweise eine Vorhersage der Wirksamkeit einer Anti-PD-L1-Therapie bzw. deren Sinnhaftigkeit möglich.
3.16.2.3 Immunsuppressive Zellen
Tregs
Regulatorische T-Zellen (Tregs) unterdrücken Effektor-T-Zellen und APCs – beim Gesunden, um den Immunstatus aufrecht zu erhalten und für Selbsttoleranz zu sorgen. Das tun sie mittels drei wesentlicher Mechanismen:
- Sie „konsumieren“ IL-2 durch ihren IL-2-Rezeptor, auch CD25.
- Sie regeln für Effektor-T-Zellen kostimulatorische Signale durch Bindung von CTLA4 und CD80/86 herunter.
- Sie sekretieren inhibitorische Zytokine wie IL-35, IL-10, TGF-b oder induzieren die Apoptose durch die Produktion von Perforin und Granzyme.
Zusätzlich zu diesen Mechanismen sorgt ein Ungleichgewicht von Tregs >> T-Zellen im TME für eine entsprechende, lokale Immunosuppression bzw. für die tumorale Evasion.
3.16.2.4 MDSC
Myeloid Derived Suppressor Cells (MDSC) sind Schlüsselmediatoren im TME, die den Immunstatus kontrollieren. Sie üben multiple Funktionen aus wie die Unterdrückung von CD8+ T-Zellen, sie sorgen für Angiogenese, Zellinvasion und für die Metastasierung von Tumorzellen. Auch sie behindern/erschweren eine ICI-Therapie.
3.16.2.5 TAMs
Je nach Umgebung in die Makrophagen migrieren, können sie in M1 oder M2 Makrophagen eingeteilt werden. M2 Makrophagen verfügen über tumorigene Eigenschaften. Diejenigen, die im/vom TME „erzogen“ worden sind, differenzieren zu spezifischen tumorassoziierten Makrophagen (Tumor Associated Macrophages), kurz TAMs. Eine erhöhte Menge an TAMS ist bei humanen Tumoren mit Metastasierung und schlechter Prognose assoziiert. In vivo lässt sich nachweisen, dass TAMs Anti-PD-1-Antikörper von der T-Zelloberfläche unmittelbar nach der Bindung des Antikörpers an die PD-1-positiven, tumorinfiltrierenden CD8+ T-Zellen abfangen, was in der Konsequenz auch eine Resistenz gegenüber einer ICI-Therapie zur Folge hat.
3.16.2.6 T-Zell-Erschöpfung (Exhaustion)
Dass T-Zellen ein tumorbekämpfendes Potenzial haben, wurde schon mehrfach dargelegt. Sie sind ausschlaggebend für eine immunologische Tumorantwort. Nun ist es so, dass eine dauerhafte Stimulation mit tumorspezifischen Neoantigenen auch erhöhte Spiegel von PD-1 auf Effektor T-Zellen induziert. Die Folge ist ein Erschöpfungs- und immunologisch ineffektiver Zustand. Hinweis: Je mehr PD-1 auf einer T-Zelle exprimiert wird, umso höher ist ja auch die Wahrscheinlichkeit, dass ein inaktivierender PD-1-Ligand daran bindet (siehe Abschnitt Programmed Cell Death Protein 1).
3.16.2.7 Der epithelial-mesenchymale Übergang (Epithelial-Mesenchymal Transition – EMT)
EMT ist der Prozess, bei dem eine Differenzierung von epithelialen Tumorzellen zu einem mesenchymalen Phänotyp stattfindet. Dieser Phänotyp verleiht den Zellen Motilität sowie invasive und metastatische Eigenschaften, u. a. durch Tumorzellplastizität. EMT und Tumorzellplastizität haben sich als Resistenzfaktoren gegenüber antitumoralen Therapien, sei es die zytotoxische oder auch die sog. zielgerichtete Therapie, erwiesen. Durch den EMT erwerben mesenchymal-ähnliche Tumore multiple Immun-Escape-Mechanismen wie:
- Resistenz gegenüber immun-mediierter Zytotoxizität durch T- und NK-Zellen
- Expression des PD-L1 auf Tumorzellen
3.16.3 Weitere Immuncheckpoints nicht-CTLA4/PD-1/PD-L1
Auf T-Zellen gibt es weitere Immuncheckpoints, wohingegen deren Liganden sich auf Tumorzellen befinden können.
3.16.3.1 Tim-3
Ein weiterer inhibitorischer Immuncheckpoint ist T-Cell Immunglobulin and Mucin 3 (Tim-3). Tim-3 findet sich üblicherweise auf T-Zellen, Makrophagen und NK-Zellen. Im Speziellen limitiert Tim-3 die Stärke und die Dauer von IFN-g produzierenden CD4+ T1 Helferzellen (Th1) und CD8+ T-Zellen. Neben seinem Einfluss auf die aktivierte T-Zellpopulation fördert Tim-3 auch die Funktionen von immunsuppressiven MDSC als auch die von tumorassoziierten dendritischen Zellen (TADC). Es stimuliert die Expansion von Tregs und MDSC im TME.
3.16.3.2 TIGIT
TIGIT (T-Cell Immunoglobulin and ITIM Domain) mit ITIM (Immunoreceptor Tyrosine-based Motif) ist ein inhibitorischer Rezeptor auf Lymphozyten, hauptsächlich auf T- und NK-Zellen zu finden. Sein Ligand ist CD155 oder auch Poliovirusrezeptor, CD112 und CD113, wobei CD155 den Hauptliganden darstellt. Die Rolle von TIGIT im TME ähnelt der PD-1/PD-L1 Interaktion tumoraler Immunosuppression. CD155 findet sich auch auf APCs.
3.16.3.3 LAG-3
LAG-3 (Lymphocyte-Activation-Gene-3), auch CD223, findet sich auf T-, NK-, B-Zellen, DCs und Tregs. LAG-3 hat drei Hauptliganden:
- MHC II Moleküle auf APCs
- Galectin-3
- LSECtin = Liver Sinosidal Endothelial Cell Lectin.
Ihre Aufgabe ist
- Kompetitive Inhibition von Antigenen und des T-Zellrezeptors
- Suppression von tumorspezifischen T-Zellantworten und die
- Inhibition von antitumoralen T-Zellantworten.
Verglichen mit anderen inhibitorischen Immuncheckpoints, scheint LAG-3 eine limitierte Funktionalität resp. eine milde Immunosuppression zu haben. Allerdings sind LAG-3+ Tregs stark immunsuppressiv. Ein hoher prozentualer Anteil an LAG-3+ Tregs im TME wird beim DLBCL als Biomarker für T-Zell-Erschöpfung gewertet.
3.16.4 Enzyme und Mikrobiom
3.16.4.1 Indolamin 2, 3-dioxygenase (IDO)
Die IDO ist ein Enzym, welches die essenzielle Aminosäure Tryptophan zu Kynurenin katabolisiert. Neben diversen Aufgaben und Funktionen ist Tryptophan essenziell für die T-Zell-Proliferation. Tumore können IDO hochregulieren, wodurch es zu einem Mangel an Tryptophan im T-Zellmilieu und damit auch zu peripherer Toleranz kommt. Verarmung an Tryptophan inhibiert die Serin-/Threoninkinase mTORC1 und aktiviert eine weitere Serin-/Threoninkinase GCN2. GCN2 ist mittlerweile als ein weiterer Immuncheckpoint realisiert. Er inhibiert z. B. Translation, und kontrolliert den Zellzyklus, indem der Übergang in die S-Phase verzögert wird. Auf T-Zellebene führt ein Tryptophanmangel zur Anergie von T-Effektorzellen. Darüber hinaus aktiviert der Metabolit Kynurenin AHR, was zur Differenzierung von immunsupprimierenden Tregs führt.
Neben den Immunescapemechanismen wie oben beschrieben moduliert die IDO auch die Level bestimmter inflammatorischer Zytokine wie IFN-g und IL-6, was zur Tumorzellproliferation führt.
3.16.4.2 Das Darmmikrobiom
Man geht mittlerweile davon aus, dass das Mikrobiom des menschlichen Darms „irgendwie“ mit diversen Erkrankungen assoziiert ist, wie beispielsweise entzündliche Darmerkrankungen, Arthritis und auch Parkinson. Das Mikrobiom kann auch eine Krebsprognose beeinflussen. Die antitumorale Immunität kann mittels des Mikrobioms über die Aktivierung von DCs im Intestinum moduliert werden. Die Migration enteraler Bakterien in das TME mit gleichzeitiger Aktivierung von Effektor T-Zellen spielt eine entscheidende Rolle bei der Auslösung einer Antitumorimmunität. Produkte enterobakterieller Flora, wie pathogenassoziierte molekulare Muster, können die Antigenpräsentation induzieren und zu einer Aktivierung von T-Zellen, DCs und Makrophagen führen.
In Übereinstimmung mit diesen Daten konnte eine Korrelation zwischen dem Ansprechen auf eine Immuntherapie und dem Vorkommen bestimmter Darmbakterien hergestellt werden. Während Spezies wie Bifidobacterium, Collinsella, Faecalibacterium, Ruminococcaceae, Akkermansia und Enterococcus angereichert im Darm von Respondern einer Anti-PD-1-Therapy gefunden wurden, fand man Spezies wie Bacteroides angereichert in non-Respondern. Bacteroides Spezies waren auch positiv assoziiert mit der Wirksamkeit einer Anti-CTLA4-Antikörpertherapie. Experimentelle Mikrobiomtransplantationen von Patienten mit gutem Ansprechen auf eine Anti-CTLA4-Antikörpertherapie in keimfreie („sterile“) Mäuse mit transplantierten Tumoren hatten eine deutlich bessere Tumorkotrolle als Mäuse mit dem Mikrobiom von non-Respondern. Ähnliche Ergebnisse gibt es auch zur Anti-PD-1-Therapie.
Die Mäuse mit dem Mikrobiom der Responder hatten eine bessere Tumorkontrolle und ein besseres Ansprechen auf die Anti-PD-1-Therapie. Dieses Ansprechen korrelierte mit einer erhöhten Anzahl antitumoraler CD8+ T-Zellen im Tumor. Die Mäuse mit dem transplantierten Mikrobiom der non-Responder sprachen entsprechend schlecht auf die Anti-PD-1-Therapie an und hatten eine hohe Dichte an Tregs im TME.
Es gibt also Hinweise, dass ein bestimmtes, mikrobiontisches Darmmilieu die Effektivität einer ICI-Therapie verstärken kann. Der präzise immunologische und metabolische Mechanismus dazu ist derzeit jedoch unbekannt. Wir befinden uns diesbezüglich noch ziemlich weit unten auf der Lernkurve.
3.17 Überwindung der ICI-Resistenzen?
Synergismen, um die Ergebnisse eine Checkpoint-Inhibitortherapie zu verbessern, sind in Anfängen bereits in der Klinik angekommen. So kann die Kombination anti-CTLA4 plus anti-PD-1 beim Melanom, Nierenzellkarzinom und beim NSCLC bei Erfüllung der Therapievoraussetzungen angewendet werden. Die Kombination von anti-PD-1 plus Antiangiogenese ist ebenfalls beim Nierenzellkarzinom zugelassen. Die Kombination von ICI mit konventioneller CTX nicht zu vergessen.
Aus den Ausführungen in den Unterkapiteln ergeben sich – theoretisch – Strategien, um Resistenzen gegenüber ICI zu umgehen bzw. aufzuheben. Es seien kursorisch genannt (siehe auch jeweiliges Unterkapitel):
- Erhöhung der Neoantigene und/oder deren Präsentation (siehe Abschnitt T-Zell-Erschöpfung)
- Aufhebung der epigenetischen Veränderungen (siehe auch Abschnitt HDACi und Abschnitt DNMTi)
- Restaurierung veränderter Signalwege oder Blockade dieser veränderten Signalwege (siehe auch Abschnitt Veränderungen von Signalwegen)
- Entwicklung von STING-Agonisten (siehe auch Abschnitt Abwesenheit von T-Zellen)
- Unterdrückung extrasomaler PD-L1- oder Entwicklung Anti-extrasomaler-PD-L1-Antikörper (siehe auch Abschnitt Exosomaler PD-L1)
- Unterdrückung von Tregs im TME (siehe auch Abschnitt Immunsuppressive Zellen)
- Selektive Unterdrückung der Makrophagen-PI3Kg in MDSC
- Unterdrückung von TAMs; potenzielle Wirkstoffklassen: CXCR4- und CSF-1R-Inhibitoren, Anti-Makrophagenrezeptor mit kollaginöser Struktur (MARCO – Macrophage Receptor with Collagenous Structure) Antikörper (siehe auch Abschnitt TAMs)
- Unterdrückung der EMT
- Schaffung eines günstigen („hot“) TME, z. B. durch CD40 Agonisten für immunkompetente Zellen
- Blockade weiterer negativ-regulatorischer Immuncheckpoints → Tim-3-, TIGIT-, LAG-3-Antagonisten
- IDO-Inhibitoren
Die Immunokologie bzw. deren therapeutische Ansätze ist ein weites, kompliziertes, aber auch interessantes Forschungsfeld. Für bisher schwer bis gar nicht behandelbare Tumore eröffnen sich neue Möglichkeiten, sicherlich aber auch unter Inkaufnahme des einen oder anderen Rückschlags.
3.17.1 Resistenzen gegen CAR-T-Zellen
CAR-T-Zellen sind ja im Unterschied zu Anti-CTLA4/PD-1/PD-L1-Antikörpern gegen ein Tumorzellantigen gerichtet. Die ersten Produkte sind gegen das CD19-Antigen auf B-Zelllymphom- und B-Leukämiezellen gerichtet. Auch unter einer CAR-T-Zelltherapie kann es zu Rezidiven auf Grund von Resistenzen kommen.
3.17.1.1 T-Zell-Faktoren
Die Antwort auf eine antitumorale Immuntherapie hängt vom Status der Immunfunktionalität ab. Ein Defekt im Immunsystem kann folglich die Prognose der Patienten verschlechtern. So ist bei einem Vergleich verschiedener CAR-T-Zellstudien die Wirksamkeit der Therapie gegen eine CLL deutlich schlechter als gegen eine B-ALL. Das wird auf Defekte des angeborenen Immunsystems der CLL-Patienten zurückgeführt. T-Zelldefekte zum Zeitpunkt der Ernte (Apharese) können dann einen Einfluss auf das Ergebnis der CAR-T-Zellproduktion haben. Expansion, Persistenz und antitumorale Zytotoxizität sind 3 Hauptcharakteristika von CAR-T-Zellen, die die therapeutische Wirksamkeit bedingen und beeinflussen. T-Zellen von Tumorpatienten können aber eine verminderte intrinsische Zytotoxizität aufweisen, wofür es aber keine leicht zugänglichen Biomarker gibt. Diese verminderte intrinsische Zytotoxizität kann auch durch ex vivo Modifikationen nicht zwingend verbessert oder aufgehoben werden.
Mit dem Alter des T-Zellpools ändert sich permanent dessen Zusammensetzung. So verschiebt sich der Anteil von undifferenzierten, naiven T-Zellen zu differenzierten Effektor-/Gedächtniszellen, denen CD28 fehlt und die nach Stimulation mit Antigenen eine verminderte Proliferationsfähigkeit aufweisen. Auch CAR-T-Zellen können der Erschöpfung unterliegen (siehe Abschnitt T-Zell-Erschöpfung). Zum einen kann eine Verminderung an Effektoren und eine gleichzeitige Erhöung der Expression inhibitorischer Rezeptoren, z. B. durch chronische Stimulation, zu dieser Dysfunktion führen. Zum anderen gibt es Hinweise aus Studien, dass sich CAR-T-Zellen spontan, antigenunabhängig zusammengruppieren (clustern) können und dadurch ein CAR-CD3z-Signal generieren, dass zur CAR-T-Zellexhaustion führt.
3.17.1.2 Tumor-Faktoren
Die tumoralen Faktoren können prinzipiell in zwei Kategorien eingeteilt werden: in Rezidive mit antigennegativer Expression und in Rezidive mit antigenpositiver Expression.
3.17.1.3 Zielantigen-negatives Rezidiv
Die Mechanismen von Zielantigen-negativen Rezidiven umfassen hauptsächlich 4 Aspekte:
- Präexistierende, Zielantigen-negative Tumorzellen (Zielantigen-negativer Subklon)
- Verminderte Expression des Zielantigens
- Mutations-, Splicevarianten oder Zelllinienwechsel-mediierter Antigenverlust
- Versagen der Ziellinienantigenpräsentation
Für diese Art der Rezidive gibt es derzeit den größten Datensatz mit anti-CD19 gerichteten CAR-T-Zellprodukten.
3.17.1.3.1 Präexistierende, Zielantigen-negative Tumorzellen – negativer Subklon
Die Wirksamkeit einer solchen Therapie hängt auch mit der Dichte des Zielantigens auf der Tumorzellmembran ab. So hat eine Forschergruppe vom Children´s Hospital of Philadelphia 628 Fälle von refraktären oder rezidivierten B-ALLs untersucht. Sie konnten mittels Durchflußzytometrie zeigen, dass 17% der Fälle prätherapeutisch CD19-negative Tumorzellen aufwiesen, definiert als ³ 1% an CD19 negativen Zellen. Verglichen mit Gesunden, zeigten 7% der Patienten eine verminderte, und 24% eine normale CD19 Expression.
3.17.1.3.2 Verminderte Expression des Zielantigens, Mutations- und Splicevarianten
Es konnten weiterhin Exon 2-Splicevarianten der mRNS für CD19 gefunden werden, die in der Konsequenz das CD19-Antigen auf den Tumorzellen verschwinden ließen. Eine Exon 5 und Exon 6 mediierte Defizienz der transmembranösen CD19-Domäne induziert ebenfalls ein CD19-negatives Rezidiv gegen eine Anti-CD19 CAR-T-Zelltherapie.
Mutationen in den Exonen 2 bis 5, wie Rastermutationen (Frameshift) in den Exonen 2, 3 oder 4, Insertionen in Exon 3 und nicht-synonyme Mutationen in Exon 4 führen zum CD19-Verlust in der Zellmembran und zu post-CD19 CAR-T-Zelltherapierezidiven.
3.17.1.3.3 Antigenverlust durch Zelllinienwechsel – Lineage Switching
Zelllinienwechsel (Lineage Switching) im Sinne einer Konversion kann ebenfalls zu CD19-regativen Rezidiven führen. Forschungsergebnisse haben gezeigt, dass Fälle von B-ALL mit Eradikation der Blasten nach einer Anti-CD19-CAR-T-Zellinfusion, mit einem CD19-negativen Rezidiv und myeloidem Phänotyp imponierten. Wie dieses Lineage Switching mechanistisch funktioniert, ist schwer nachvollziehbar. Es existieren hierzu aber 2 Mainstreamtheorien:
- Eine arzneimittelindizierte Reprogrammierung der originären Tumorstammzellen gefolgt von der Expansion unterschiedlicher, phänotypischer Klone von Tumorzellen während/unter der Therapie (vielleicht im Sinne von Selektionsdruck).
- Post-translationals, defiziente Ausreifung und Translokation von CD19
3.17.1.3.4 Maskierung des Antigenepitops (Epitope Masking)
Die Epitopenmaskierung wurde durch einen sehr seltenen und sehr unglücklichen Zufall entdeckt und stellt ein potenzielles Escapephänomen dar.
Ein 20-jähriger Mann wurde nach 3 chemotherapeutisch behandelten Rezidiven einer experimentellen CAR-T-Zelltherapie zugeführt. Am Tag 28 nach der CAR-T-Zellinfusion befand er sich in kompletter Remission. Neun Monate später erlitt er ein CD19-negatives Rezidiv. Im Rahmen der Aufarbeitung des Rezidivs wurden zunächst die erwähnten CD19-Mutationen und Splicevarianten ausgeschlossen. Es stellte sich heraus, dass der Anti-CD19-CAR während der gentechnischen Aufarbeitung in eine einzelne leukämische B-Zelle eingearbeitet wurde. Dieser Tumorklon wurde dann mit den CAR-T-Zellen dem Patienten retransfundiert, wo er letzendlich auch expandierte. Das führte zum Progess mit anschließendem Tod des Patienten. Der Progress resultierte daraus, dass das CAR-Molekül dem CD19-Antigen benachbart war, sodass das eigentliche Epitop wie von einem Regenschirm abgedeckt – maskiert – war und von den Anti-CD19-CAR-T-Zellen nicht mehr erreicht werden konnte.
3.17.1.4 Zielantigen-positives Rezidiv
Eine Vermutung der Ursache eines Zielantigen-positiven Rezidivs ist eine kurze Persitenz der CAR-T-Zellen im Patienten. Die Gründe dafür sind komplex und auf individueller Basis kaum zu ermitteln. Eine grobqualitative Beschreibung ist die Annahme, dass ein immunsupressives TME eine Rolle spielt. Wie erwähnt sind CAR-T-Zellen ja chimäre, da bei den derzeitigen Produkten der scFv-Teil murinen Ursprungs ist. Bei einer Untergruppe rezidivierter Patienten war der CAR-T-Zellverlust mit einer Anti-CAR-T-Zellimmunantwort gegen eben diese Epitope verknüpft, bildlich gesprochen über eine Art Anti-scFv-CAR-T-Zellantikörper (Hypothese).
Nun üben CAR-T-Zellen ihren antitumoralen Effekt nicht allein durch die Antigenerkennung aus, sondern durch Induktion der Apoptose. Apoptoseinduzierende (extrazelluläre) Signale umfassen (u. a.):
- TRAIL = TNF-related apoptosis-inducing ligand
- FasL = Fas-Ligand; der Fas-Ligand (CD95-Ligand, CD95L) ist ein zur TNF-Familie gehörendes Glykoprotein der Zelloberfläche, das nach Wechselwirkung mit dem Fas-Rezeptor (FasR, kurz Fas oder CD95 oder Fas/Apo-1 oder „Todesrezeptor“) den programmierten Zelltod (Apoptose) auslöst und darüber hinaus eine Ca2+-unabhängige Zytotoxizität bewirkt.
- Zytokine wie IFN-g
Experimente haben gezeigt, dass bei normal funktionierenden CAR-T-Zellen, gemessen an deren Zytokinsekretion, TRAIL-Inhibitoren (vom Tumor synthetisiert) deren Zytotoxizität unterdrücken kann. Der TRAIL-Signalweg kann in Tumorzellen auch komplett verloren gehen (Anti-Apoptose).
Wie bei den Resistenzen gegenüber ICI beschrieben, kann es, basierend auf Mutationen, zu Veränderungen von Signalwegen kommen (siehe Abschnitt Resistenzen gegen monoklonale Antikörper). Erwähnt sei auch hier der PTEN-Loss und und Loss-of-Function von JAK1 oder -2. Es ist aber unklar, ob die Resistenzmechanismen der Immuncheckpoint-Inhibitoren so auf die CAR-T-Resistenzen übertragbar sind.
3.17.2 Überwindung der CAR-T-Zellresistenzen?
Wie bei der Umgebung von ICI-Resistenzen ergeben sich auch bei den CAR-T-Zellen, zumindest theoretisch, Strategien, die Resistenzen zu überwinden.
3.17.2.1 Verbessertes CAR-T-Zelldesign (Engineering)
Erste Ansätze zur Optimierung von CAR-T-Zellen sind weiter oben beschrieben (siehe Abschnitt Hämatologie in Fahrt). Weitere, denkbare Modifikationen wären:
- Voll humane CARs, um eine Immunantworgt gegen das derzeit murine scFv-Fragment auszuschließen
- Weitere ko-stimulatorische Moleküle
- Modifikation von CAR-T-Zellen, sodass eine geringere Antigendichte benötigt wird, um sie zu stimulieren
- Fähigkeit, mehr spezifischer Zytokine im TME auszuschütten, beispielsweise IL-2 und IL-12 (CAVE: Toxizität?)
- „Bewaffnete“ CAR-T-Zellen. Diese könnten nach Aktivierung im TME weitere Zytokine ausschütten oder Substanzen, die das immunsuppressive TME in ein proinflammatorisches TME überführen. In diesem Zusammenhang auch
- Steigerung der Apoptoseempfindlichkeit z. B. durch Ausschüttung proapoptotischer Moleküle im TME oder Unterdrückung antiapoptotischer Faktoren
- CAR-T-Zellen, die im TME PD-1-Inhibitoren sekretieren
- Gleichzeitige oder sequenzielle Infusion von CAR-T-Zellen mit unterschiedlicher Antigenspezifität. Dafür muss ein Tumor aber mehr als ein Antigen aufweisen.
- Multi-Targeted-CAR-T-Zellen – Verwendung eines Vektors, der für 2 oder 3 unterschiedliche CARs einer einzelnen T-Zelle kodiert (bi-, tricistronische CAR)
- Allogene CAR-T-Zellen gesunder Spender (CAVE: potenzielle GvHD-Reaktionen)
- Kombinationstherapien mit ICI, Radiotherapie, oder weiteren Inhibitoren wie beispielsweise Treg selektive PI3K-Inhibitoren
- Knock-down des für die Kodierung von PD-1 notwendigen PDCD1 Gens
Ansonsten gelten auch die Überlegungen zur Optimierung des TME wie bei der Resistenz gegenüber ICI dargelegt.
Die Immunonkologie (IO) mit ICIs und CAR-T-Zellen hat die Landschaft der Krebstherapie dramatisch verändert. Trotz beeindruckenden Ansprechens werden aber auch hier Resistenzen beobachtet. Das Verständnis dieser Mechanismen führt zu neuen Einblicken in das Immunsystem und befeuert die Forschung, diese Resistenzen zu umgehen und/oder zu überwinden. Wie erfolgreich und humankompatibel das sein wird, zeigt die Zukunft. Die einsetzbaren IO Produkte basieren auf wissenschaftlich (hochpreisiger) Plattformtechnologie.
Literatur
- Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumors: Revised RECIST guideline (version 1.1). European Journal of Cancer 45: 228–247
- Barth J (2008) Zytostatikaherstellung in der Apotheke; einschließl. 6 Akt. Lfg. 2017, Deutscher Apotheker Verlag, Stuttgart
- Paulsen und Ferguson-Smith (2001) J Pathol 195: 97
- Force T, Krause DS, Van Etten RA (2007) Molecular mechanisms of cardiotoxicity of kinase inhibition. Nat Rev Cancer 7: 332–344
- Parkin DM (2006) Int J Cancer 118: 3030–3044
- zur Hausen et al (2009) Virology 384: 260–265
- Klussmann et al (2001) Cancer 92: 2875–2884
- Impfbrief, Dezember 2011, 56. Ausgabe
- WHO Information centre, summary report HPV, Sept. 2010, https://www.unav.edu/documents/16089811/16216616/HPVReport2010.pdf (aufgerufen am 15.3.2021)
- Fachinformation Cervarix 2013
- Fachinformation Gardasil 2013
- http://www.who.int/medicines/services/inn/en/ und http://www.who.int/medicines/services/inn/meetings/en/index.html (beide aufgerufen am 15.3.2021)
- O’Shaughnessy J, Blackwell K, Burstein H et al (2008) A randomized study of Lapatinib (Tykerb/Tyverb.) in combination with Trastuzumab versus Lapatinib monotherapy in heavily pretreated HER2+ metastatic breast cancer progressing on Trastuzumab therapy. ASCO-Meeting 2008, Vortrag
- Zeller/zur Hausen (1995) Onkologie; einschließlich 1. Erg. Lfg. 1996, ecomed Medizin, Landsberg
4. Wechselwirkungen von Zytostatika mit anderen Arzneimitteln
Irene Krämer
Unter Wechselwirkungen oder Interaktionen versteht man die Beeinflussung der Wirkung eines Arzneimittels durch ein anderes gleichzeitig angewendetes Arzneimittel. Die Wechselwirkung kann zu einer Wirkungsverstärkung oder einer Wirkungsabschwächung eines Arzneimittels führen. Die Wahrscheinlichkeit der Wechselwirkung wächst exponenziell mit der Anzahl der eingesetzten Arzneimittel. In der Regel werden dabei immer zwei Arzneimittel als Interaktionspartner betrachtet. Zu den Wechselwirkungen von Dreierkombinationen oder anderen Mehrfachkombinationen gibt es kaum Untersuchungen. Onkologische Patienten haben ein hohes Risiko für Wechselwirkungen, da sie durchschnittlich zehn verschiedene Arzneimittel einnehmen. Insbesondere ältere Patienten erhalten eine Poly-/Multimedikation wegen Komorbiditäten. Von den hohen Raten der Wechselwirkungen ist nur ein geringer Teil (ca. 20 %) klinisch relevant und mit schwerwiegenden unerwünschten Wirkungen verbunden. Allerdings führen diese dann häufig zu Symptomen und zur Krankenhausaufnahme. Klinisch relevante Wechselwirkungen treten insbesondere dann auf, wenn Arzneimittel mit geringer therapeutischer Breite, also einem schmalen Dosisbereich zwischen therapeutischer und toxischer Wirkung, gleichzeitig angewendet werden. Antineoplastische Arzneimittel gehören zu den Arzneimitteln mit geringer therapeutischer Breite. Da sie in der Regel in Kombination und zusätzlich weitere Arzneimittel als Begleit- und Supportivtherapie und möglicherweise noch weitere Arzneimittel für andere Erkrankungen des Patienten gegeben werden, entsteht ein hohes Interaktionspotenzial. Neben den Zytostatika und Supportivtherapeutika (insbesondere Antibiotika, Antimykotika, Schmerzmittel, Antikonvulsiva) werden nachfolgend auch bei Tumorpatienten und Blutstammzelltransplantierten häufig eingesetzte sonstige Arzneimittel betrachtet (z. B. Antihypertonika, Schilddrüsenhormone, Immunsuppressiva).
Bezüglich des Interaktionsmechanismus werden pharmakokinetische (gegenseitige Beeinflussung der Konzentrationen) und pharmakodynamische (gegenseitige Beeinflussung der Wirkung) Interaktionen unterschieden.
Pharmakodynamische Interaktionen führen durch gleichartige (additiv oder synergistisch) oder gegensätzliche Wirkung (antagonistisch) an der gleichen Zielstruktur zu Effektveränderungen. Folge können einerseits Wirkungsverstärkungen bis toxische Reaktionen oder andererseits Wirkungsabschwächungen bis hin zum Wirkungsverlust sein. In der Tumortherapie soll die Kombinationschemotherapie eine Wirkungsverstärkung herbeiführen und die Resistenzentwicklung der Tumorzellen vermeiden. Mit der Applikation von Folinsäure/Calciumfolinat kann die Wirkung von Fluorouracil verstärkt und von Methotrexat verhindert werden.
Pharmakokinetische Interaktionen können insbesondere bei der Arzneistoffaufnahme (= Resorption), der Verstoffwechselung (= Metabolismus) in der Leber und der Ausscheidung der Arzneimittel über Niere und Darm auftreten.
Pharmakokinetik nach oraler Applikation
- Freisetzung
- Aufnahme / Resorption
- Verteilung
- Verstoffwechselung / Metabolismus
- Ausscheidung
4.1 Wechselwirkungen bei der Absorption
Wechselwirkungen bei der Absorption spielen insbesondere bei der oralen Applikation eine Rolle. Während in der Vergangenheit nur wenige klassische Zytostatika oral angewendet wurden, spielt die orale Therapie mit den neuen oralen Antitumortherapeutika heute eine bedeutende Rolle. Bei den modernen oralen Antitumortherapeutika handelt sich um Arzneistoffe, die unterschiedliche Kinasen (z.B. Tyrosinkinasen, BRAF-Kinase, MEK-Kinasen) oder andere Zielstrukturen (z.B. Histon-Desacetylase, Poly(ADP-ribose)-Polymerase) hemmen. Nach der Einnahme muss der Wirkstoff aus dem Arzneimittel freigesetzt und über die Schleimhäute des Gastrointestinaltraktes (GIT) in den Blutkreislauf aufgenommen werden. Die Freisetzung und die Absorption sind u. a. vom Füllungszustand des Magens abhängig. Manche Arzneistoffe werden besser bei Nüchterneinnahme (z. B. Penicillin V), andere besser bei Einnahme mit einer Mahlzeit (z. B. Posaconazol-Suspension, Ciclosporin) resorbiert. Ein wesentlicher Faktor dafür ist die Löslichkeit des Arzneistoffs im GI-Trakt, die ihrerseits auch wieder pH-Wert abhängig ist. Der pH-Wert nimmt von extrem sauer im Magen über den oberen Dünndarm bis in den unteren Dünndarm immer mehr ab. Bei pH-abhängiger Resorption kann die gleichzeitige Anwendung von Protonenpumpenhemmern, H2-Blockern oder Antazida die Absorptionsrate verändern. Protonenpumpenhemmer führen zur andauernden Hemmung der Magensäuresekretion und bedingen damit eine pH-Wert Erhöhung und Resorptionsverminderung von oral applizierten Kinase-Inhibitoren. Klinisch relevant ist diese Wechselwirkung bei Axitinib, Bosutinib, Ceritinib, Dacomitinib, Dasatinib, Erlotinib und Pazopanib. Die Kombination mit Protonenpumpenhemmern soll möglichst vermieden werden. Ersatzweise kann eine Antacidatherapie mit Al-Mg-haltigen Antacida erfolgen. Bei Ceritinib, Dacomitinib und Erlotinib können H2-Blocker zeitversetzt, d.h. 2 Stunden oder 10 Stunden nach den Tiniben, eingenommen werden. Ausführliche Informationen zum richtigen Zeitpunkt der Einnahme der Zytostatika in Bezug auf die Mahlzeit finden sich in Teil Durchführung Abschnitt Orale Gabe.
Durch Komplexbildung von Arzneistoffen wie Tetrazyklinen, Gyrasehemmern und Bisphosphonaten mit Metallionen, wie Calcium-, Magnesium- und Eisenionen, sinkt deren Resorptionsquote klinisch relevant. Die genannten Wirkstoffe dürfen daher nicht zusammen mit Mineralpräparaten oder Milchprodukten eingenommen werden (wegen des Calciumgehalts). Zur Vermeidung der Interaktion soll zwischen den Einnahmen ein Intervall von mindestens 2 Stunden eingehalten werden.
Wechselwirkungen von oral applizierten Zytostatika mit anderen oral applizierten Arzneimitteln können auch aus der Hemmung/Induktion von Zytochrom P-450 Enzymen und Transportersystemen (insbesondere P-Glykoprotein (PGP)) im Darmtrakt resultieren (siehe auch Abschnitt Wechselwirkungen beim Metabolismus und Abschnitt Wechselwirkungen bei der Ausscheidung).
Niedrig dosierte, an Nahrungsmittel adsorbierende Arzneistoffe wie L-Thyroxin (Mikrogramm!) müssen nüchtern eingenommen werden, um eine ausreichende Resorption zu gewährleisten. Der Abstand zur nächsten Mahlzeit soll 30–60 Minuten betragen.
Die Resorption kann auch durch veränderte Motilität (z. B. Diarrhö, Motilitätshemmer, Prokinetika), eine veränderte Darmflora und Schleimhautläsionen beeinflusst werden, was auch bei oraler Zytostatikatherapie zu beachten ist.
Arzneimittelwechselwirkungen bei der Absorption betreffen die orale Applikation. Wechselwirkungen bei der Verteilung, Verstoffwechselung/Metabolismus und Ausscheidung sind unabhängig von der Art der Applikation (z. B. intravenös, oral). Das Risiko von Wechselwirkungen besteht insbesondere, wenn neue Arzneimittel angesetzt oder Arzneimittel einer bestehenden Medikation abgesetzt werden.
4.2 Wechselwirkungen bei der Verteilung
Unter Verteilung versteht man den Transport mit dem Blut zu den Organen und Geweben. Viele Arzneistoffe zirkulieren gebunden an Plasmaproteine. Die Verdrängung aus der Plasmaeiweißbindung und die daraus resultierende Wirkungssteigerung kann bei Methotrexatdosierungen ab 25 mg/Woche und der gleichzeitigen Gabe von nichtsteroidalen Antirheumatika (NSAR) zu klinisch relevanten toxischen Effekten, wie verstärkter Neutropenie, Mukositis und Nierenschädigungen führen.
4.3 Wechselwirkungen beim Metabolismus
In der Leber werden die Arzneistoffe insbesondere über das Zytochrom-P-450-Enzymsystem metabolisiert, d. h. chemisch verändert. In der Regel führen die chemischen Reaktionen zu verbesserter Wasserlöslichkeit und Ausscheidung über die Niere. Andererseits werden auch Arzneistoffe in der Leber enzymatisch in ihre Wirkform überführt (z. B. Cyclophosphamid). Die wichtigsten Zytochrom-P-450-Enzymsubtypen sind das CYP1A2, 3A4, 2D6, 2C9 und 2C19. Der am häufigsten vorkommende Subtyp ist CYP3A4, durch den etwa 50 % aller Arzneistoffe metabolisiert werden. Weitere 25 % der Arzneimittel werden durch CYP2D6 metabolisiert. Für Letzteres gibt es einen genetischen Polymorphismus, d. h. manche Menschen produzieren viel Enzym und sind Schnellmetabolisierer, andere produzieren wenig und sind sogenannte Langsammetabolisierer. Die Variabilität beträgt Faktor 1000. Die unterschiedlichen Metabolisierungsraten erhöhen das Risiko für eine Arzneimittelinteraktion. Manche Arzneistoffe werden über mehrere Subtypen von CYP 450 metabolisiert. Dies ist vorteilhaft, wenn ein Eliminationsweg blockiert ist und andere Eliminationswege genutzt werden können.
Arzneistoffe, die über die CYP-Enzyme interagieren, werden unterteilt in Substrate, Inhibitoren und Induktoren:
Substrate interagieren bei den polymorphen Enzymen CYP2D6, 2C9 und 2C19. Die Konkurrenz um das gleiche Enzym kann zu erhöhten Konzentrationen des weniger metabolisierten Arzneistoffs führen.
Inhibitoren können einzelne oder alle CYP-Enzyme hemmen, wodurch bei gleichzeitiger Anwendung eines Substrats dessen Konzentration steigt und der Effekt verstärkt wird. Es steigt das Risiko für toxische Wirkungen, ggf. muss die Dosis reduziert werden. Die Interaktion tritt als Konkurrenzreaktion unmittelbar ein.
Induktoren führen zur vermehrten Bildung von CYP-Enzymen. Bei gleichzeitiger Anwendung von Substraten werden diese schneller metabolisiert und die Konzentration sinkt schneller ab, ggf. muss die Dosis erhöht werden. Da die Neusynthese der Enzyme einige Tage Zeit beansprucht, setzt die Interaktion mit neu angesetzten Induktoren erst nach einigen Tagen ein.
Manche Arzneistoffe bilden Substrate und Inhibitoren oder Induktoren am gleichen Enzymsubtyp.
| Zytochrom-P-450-Subtyp | Substrat | Inhibitor | Induktor |
| CYP1A2 | Bortezomib, Erlotinib | Gyrasehemmer (=Chinolone) | Johanniskraut, Tabak |
| CYP2D6 | Doxorubicin, Tamoxifen | Abirateron, Amiodaron, Fluoxetin/Paroxetin, Gefitinib, Imatinib, Nilotinib, Panobinostat, Pazopanib | – |
| CYP2C9 | Capecitabin, Imatinib | Capecitabin, Azole (Fluconazol, Voriconazol), Phenytoin | Aprepitant, Phenobarbital, Johanniskraut |
| CYP2C19 | Bortezomib, Cyclophosphamid | Azole (Fluconazol, Voriconazol), Omeprazol/Esomeprazol | Carbamazepin, Phenobarbital, Phenytoin |
| CYP3A4 | Abirateron, Afatinib, Abemaciclib, Axitinib, Bosutinib, Brigatinib, Cabozantinib, Ceritinib, Cobimetinib, Crizotinib, Cycloposphamid, Cytarabin, Dabrafenib, Dasatinib, Doxorubicin, Docetaxel, Entrectinib, Erlotinib, Etoposid, Gefitinib, Gilteritinib, Ibrutinib, Idelalisib, Ifosfamid, Irinotecan, Imatinib, Ivosidenib, Ixazomib, Lapatinib, Larotrectinib, Lorlatinib, Midostaurin, Nilotinib, Olaparib, Osimertinib, Paclitaxel, Palbociclib, Panobinostat, Ponatinib, Pazopanib, Ruxolitinib, Sonidegib, Sorafenib, Sunitinib, Tamoxifen, Vemurafenib, Venetoclax, Vinca-Alkaloide, Vismodegib Atorvastatin/Simvastatin, Ciclosporin, Cumarine, Dronedaron, Fentanyl | Aprepitant/Netupitant, Azole (Fluconazol, Ketoconazol, Posaconazol, Voriconazol), Ceritinib, Ciprofloxacin Ciclosporin, Crizotinib, Dasatinib, Clarithromycin, Erythromycin, Erlotinib, Gefitinib, Grapefruitsaft, Ibrutinib, Idelalisib, Imatinib, Isoniazid, Lapatinib, Larotrectinib, Letermovir, Nilotinib, Palbociclib, Ponatinib, Ribociclib, Rucaparib | Brigatinib, Carbamazepin, Dabrafenib, Dexamethason, Ivosedinib, Johanniskraut, Lorlatinib, Midostaurin, Osimertinib, Phenobarbital, Phenytoin, Rifampicin, Vemurafenib |
4.4 Wechselwirkungen bei der Ausscheidung
Die Ausscheidung der Arzneistoffe und/oder ihrer Metaboliten erfolgt abhängig von den physikalisch-chemischen Eigenschaften über die Galle oder die Niere. Typischerweise werden Substanzen mit dem Molekulargewicht > 500 Dalton über die Galle ausgeschieden. Unter Umständen können Glukuronsäurereste im Darm wieder abgespalten werden und die Substanz wird wieder absorbiert (enterohepatischer Kreislauf).
Die Ausscheidung über die Niere erfolgt überwiegend durch glomeruläre Filtration. Eine reduzierte glomeruläre Filtration kann zur verminderten Ausscheidung und zur Kumulation führen. So können NSAR über die Hemmung der Prostaglandinsynthese zu reduzierter Nierenfunktion und damit einer verminderten Ausscheidung von Methotrexat und Ciclosporin führen. Methotrexat wird zudem aktiv sezerniert in der Niere. Die renale Sekretion kann durch andere Wirkstoffe, die auch über organische Anionentransporter (OAT) in den proximalen Tubuluszellen sezerniert werden, vermindert werden. Vorrangig zu nennen sind hier nicht-steroidale Antiphlogistika (NSAR), Penicillinderivate, Probenecid und Protonenpumpenhemmer. Bei einer Hochdosis-MTX-Therapie müssen die Protonenpumpenhemmer durch H2-Blocker ersetzt werden, um eine ausreichende Ausscheidung zu gewährleisten. Wechselwirkungen bei der Ausscheidung müssen besonders bei onkologischen Patienten mit reduzierter Nierenfunktion, die durch die Erkrankung oder durch die Chemotherapie bedingt sein kann, beachtet werden.
4.5 Wechselwirkungen an aktiven Transportsystemen
Aktive Transportsysteme können die aktive Aufnahme oder den aktiven Auswärtstransport von Substanzen in Zellen bewirken. Sie finden sich membrangebunden insbesondere an Geweben mit Barrierefunktion wie dem Intestinaltrakt, Leberzellen, Tubuluszellen der Niere und der Bluthirnschranke. Die Transportsysteme benötigen ATP als Energiequelle. Wegen der Bindung von ATP werden sie als ATP-Binding-Casette- oder ABC-Transporter bezeichnet. Für Arzneistoffe ist P-Gykoprotein (Pgp) der bedeutendste Transporter. Pgp transportiert Arzneistoffe aus der Zelle heraus und hemmt damit auch die intestinale Aufnahme von seinen Substraten. Dazu gehören viele Zytostatika, Immunsuppressiva und Antibiotika. Viele Arzneistoffe, die über CYP3A4 verstoffwechselt werden, werden auch über Pgp transportiert. Die Aktivität des Transportproteins kann durch Induktoren (z. B. Rifampicin) verstärkt und durch Inhibitoren (z. B. Ciclosporin) verringert werden. Darin besteht das Risiko der Wechselwirkung. Wird beispielsweise Docetaxel oder Paclitaxel gleichzeitig mit einem Pgp-Inhibitor appliziert, steigt die Bioverfügbarkeit und dadurch das Nebenwirkungsrisiko der Zytostatika. Werden die Zytostatika in Kombination mit einem Induktor angewandt, sinkt die Bioverfügbarkeit und die Wirkung.
Auf die Aktivität des Pgp wird auch die Resistenz von Tumorzellen gegen Anthrazykline, Paclitaxel und Vinca-Alkaloide zurückgeführt. Die Transportproteine pumpen die Zytostatika aus der Zelle und machen sie damit unempfindlich. Das kodierende Gen wird wegen dieser Wirkung als Multidrug-Resistance (MDR1)-Gen bezeichnet.
| Pgp-Substrat | Pgp-Inhibitor | Pgp-Induktor |
| Azol-Antimykotika Dabrafenib Dactinomycin Daunorubicin DAOK (Apixaban, Edoxaban, Rivaroxaban) Doxorubicin Docetaxel Erlotinib Etoposid Imatinib Irinotecan Lapatinib Mitomycin Mitoxantron Nilotinib Paclitaxel Pazopanib Sorafenib Sunitinib Vinca-Alkaloide | Amiodaron Azol-Antimykotika Clarithromycin Ciclosporin Eythromycin Grapefruitsaft Lapatinib Midostaurin Omeprazol Pantoprazol Simvastatin Sorafenib Tacrolimus Venetoclax Verapamil | Carbamazepin Dabrafenib Ivosedinib Nilotinib Johanniskraut Rifampicin |
4.6 Spezifische Wechselwirkungen von klassischen Zytostatika
In der nachfolgenden Tabelle werden ausgewählte Interaktionen von Zytostatika mit anderen Arzneimitteln, resultierende Effekte, der wahrscheinliche Wirkungsmechanismus und mögliche Handlungsoptionen dargestellt.
| Zytostatikum | Weitere Arzneimittel | Effekt | Mechanismus der Interaktion | Handlungsoption |
| Aldesleukin | Kortikosteroide | ↓ Antitumorwirkung von Aldesleukin | Dexamethason hemmt die von Aldesleukin induzierte Freisetzung von Tumornekrosefaktor | Kombination möglichst |
| Busulfan | CYP3A4-Inhibitoren | ↑ Toxizität von Busulfan, z. B. venoocclusive disease, pulmonale Fibrose | Hemmung des CYP3A4-Metabolismus von Busulfan | Monitoring der Busulfanspiegel, Kombination möglichst vermeiden |
| Capecitabin | Phenprocoumon | ↑ Blutungsgefahr | Hemmung des CYP2C9-Metabolismus von Phenprocoumon durch Capecitabin als CYP2C9-Inhibitor | engmaschige INR-Kontrolle und ggf. Dosisanpassung von Phenprocoumon |
| Brivudin | ↑ Konzentration und Toxizität von Capecitabin/5-FU | Hemmung der Dihydropyrimidindehydrogenase | Kontraindiziert Mindestens 4 Wochen Abstand | |
| Cisplatin | Methotrexat, Bleomycin Ifosfamid | ↑ Toxizität der Kombinationspartner | reduzierte GFR durch Cisplatin bedingte Nephrotoxizität, Reduktion der renalen Ausscheidung | GFR kontrollieren und ggf. Dosierung der Kombinationspartner an Nierenfunktion anpassen |
| Cyclophosphamid | Trastuzumab | ↑ kardiale Toxizität | additive Effekte (?) | Monitoring der kardialen Funktion |
| Docetaxel | CYP3A4-Inhibitoren Pgp-Inhibitoren Doxorubicin | ↑ Risiko der Myelosuppression | Hemmung des CYP3A4-Metabolismus von Docetaxel | Monitoring der Myelosuppression, peripheren Neuropathie usw. |
| Doxorubicin | Paclitaxel | ↑ Risiko der Neutropenie, Mukositis, Kardiomyopathie | reduzierte Doxorubicin-Clearance | Doxorubicin vor Paclitaxel applizieren |
| Etoposid | CYP3A4-Inhibitoren Pgp-Inhibitoren | ↑ Risiko der Myelosuppression | Hemmung des CYP3A4-Metabolismus von Etoposid | Monitoring der Myelosuppression |
| Irinotecan | CYP3A4-Inhibitoren | ↑ erhöhte Konzentration und Toxizität des aktiven Metaboliten SN38 | Hemmung des CYP3A4-Metabolismus von Irinotecan | Dosisreduktion von Irinotecan |
| Irinotecan | CYP3A4-Induktoren | ↓ Antitumorwirkung von Irinotecan | intensiver CYP3A4-Metabolismus von Irinotecan | Absetzen der CYP3A4-Induktoren oder Dosiserhöhung von Irinotecan |
| Mercaptopurin | Allopurinol | ↑ erhöhte Konzentration und Toxizität von Mercaptopurin | Hemmung des Abbaus durch Xanthinoxidase | Allopurinol absetzen oder Dosisreduktion von Mercaptopurin um 75% |
| Methotrexat (insbesondere Hochdosis) | NSAR, Ciclosporin, Ciprofloxacin, hochdosierte Penicilline, Piperacillin/Tazobactam, Probenecid, Protonen- pumpen-Inhibitoren Sulfamethoxazol/Trimethoprim | ↑ Toxizität von Methotrexat | Verminderung der renalen Ausscheidung | keine Applikation von NSAR innerhalb von 10 Tagen der HD-MTX-Therapie engmaschiges Monitoring der MTX Spiegel, Penicilline durch Cephalosporine ersetzen, PPI durch H2-Blocker ersetzen |
| Paclitaxel | Trastuzumab | ↑ kardiale Toxizität | additive Effekte (?) | Monitoring der kardialen Funktion |
| Procarbazin | Tyraminhaltige Lebensmittel, MAO-Hemmer: Antidepressiva, Linezolid, Morphin, Tramadol | Serotoninsyndrom | Procarbazin wirkt selbst auch als MAO-Hemmer und hemmt den Abbau von Katecholaminen | Kombination möglichst vermeiden, Monitoring bei hypertensiven Patienten |
| Procarbazin | CYP3A4 induzierende Antikonvulsiva | ↑ Risiko für Hypersensitivitätsreaktionen bei Patienten mit ZNS-Tumoren | verstärkte Bildung von ursächlichen Procarbazin-Metaboliten | Umstellung des Antikonvulsivums |
| Tamoxifen | Fluoxetin, Paroxetin | ↓ Antitumorwirkung von Tamoxifen durch CYP2D6 Inhibition | Reduzierte Metabolisierung von Tamoxifen (pro-drug) zum aktiven Metaboliten | Kontraindiziert Andere Antidepressiva einsetzen: Sertralin, Venlafaxin |
| Temsirolimus | CYP3A4-Induktoren | ↓ Antitumorwirkung von Temsirolimus | intensiver CYP3A4-Metabolismus von Temsirolimus und dessen Metaboliten Sirolimus | Absetzen der CYP3A4-Induktoren oder Dosiserhöhung von Temsirolimus |
| Vinblastin | Erythromycin | ↑ Risiko für Neurotoxizität, Ileus, Knochenmarktoxizität | Hemmung des CYP3A4-Metabolismus und Pgp | Kombination vermeiden |
| Vincristin | CYP3A4-Inhibitoren (wie Azol-Antimykotika, Isoniazid, Letermovir) | ↑ erhöhte Konzentration und Risiko der peripheren Neuropathie | Hemmung des CYP3A4-Metabolismus und Pgp | Kombination möglichst vermeiden, Monitoring der Neurotoxizität, ggf. Dosisreduktion von Vincristin |
4.7 Spezifische Wechselwirkungen von neuen oralen Antitumortherapeutika
Viele der neuen oralen Antitumortherapeutika werden als Substrate über das CYP-Enzymsystem metabolisiert und sind gleichzeitig Inhibitoren oder Induktoren verschiedener CYP-Enzyme. Ähnliches gilt für Pgp und Antitumortherapeutika Sie sind Substrate, Inhibitoren und Induktoren des Pgp (siehe dazu Tabellen oberhalb). In der nachfolgenden Tabelle werden ausgewählte Interaktionen und die resultierenden Effekte/Maßnahmen dargestellt.
| Neue orale Antitumortherapeutika ATT) | Interaktionspartner | Effekt und Maßnahme |
| Abirateron Ixazomib | Carbamazepin Johanniskraut Phenytoin Rifampicin | Plasmakonzentration der ATT verringert durch CYP3A4-Induktion Kombination möglichst vermeiden. |
| Axitinib, Bosutinib, Erlotinib, Ibrutinib, Nilotinib | Ciprofloxacin | Plasmakonzentration der ATT erhöht durch CYP3A4-Inhibition Kombination möglichst vermeiden |
| Axitinib, Bosutinib, Brigatinib, Ceritinib, Crizotinib, Dabrafenib, Dasatinib, Entrectinib, Erlotinib, Gefitinib, Gilteritinib, Ibrutinib, Idelalisib, Imatinib, Ivosidenib, Larotrectinib, Lorlatinib, Midostaurin, Nilotinib, Osimertinib Ponatinib, Ruxolitinib, Venetoclax | Aprepitant, Netupitant | Plasmakonzentration der ATT erhöht durch CYP3A4-Inhibition Engmaschiges Monitoring der Toxizität |
| Azol-Antimykotika, Clarithromycin, Isoniazid, Letermovir | Plasmakonzentration der ATT erhöht durch CYP3A4-Inhibition Kombination möglichst vermeiden | |
| Carbamazepin, Johanniskraut, Phenytoin, Rifampicin | Plasmakonzentration der ATT verringert durch CYP3A4-Induktion Kontraindiziert | |
| Dexamethason | Plasmakonzentration der ATT verringert durch CYP3A4-Induktion. Dexamethason: Plasmakonzentration verringert durch Dabrafenib, Ivosidenib (CYP3A4-Induktion), erhöht durch Ceritinib (CYP3A4-Inhibition). Kombination möglichst vermeiden | |
| Brigatinib, Dabrafenib, Lorlatinib, Osimertinib | Cumarine (Phenprocoumon, Warfarin) | Plasmakonzentration der Cumarine verringert durch CYP3A4-Induktion. Erhöhtes thromboembolisches Risiko. Engmaschige INR Kontrolle, ggf. Dosisanpassung |
| Ceritinib, Dasatinib, Erlotinib, Gefitinib, Ibrutinib, Idelalisib, Imatinib, Nilotinib, Ponatinib, Venetoclax | Cumarine (Phenprocoumon, Warfarin) | Plasmakonzentration der Cumarine erhöht durch CYP3A4-Inhibition. Erhöhtes Blutungsrisiko. Engmaschige INR Kontrolle, ggf. Dosisanpassung |
| Dabrafenib, Ivosedinib, Nilotinib | Apixaban, Rivaroxaban (DOAK) | Plasmakonzentration der DOAKs verringert durch PGP Induktion Kombination möglichst vermeiden |
| Midostaurin, Venetoclac | Apixaban, Edoxaban, Rivaroxaban (DOAK) | Plasmakonzentration der DOAKs erhöht durch PGP Inhibition Kombination möglichst vermeiden |
| Dasatinib, Ibrutinib, Ponatinib, | Apixaban, Edoxaban, Rivaroxaban (DOAK) | Additives Blutungsrisiko Kombination möglichst vermeiden |
| Ceritinib, Dasatinib, Idelalisib, Imatinib, Larotrectinib, Nilotinib, Venetoclax | Atorvastatin, Simvastatin | Plasmakonzentration der Statine erhöht durch CYP3A4-Inhibition Kombination möglichst vermeiden |
| Arsentrioxid, Ceritinib, Crizotinib, Dabrafenib, Entrectinib, Gilteritinib, Ivosidenib, Midostaurin, Panobinostat | Chinolone, Ivabradin, Makrolide | QT-Zeit-Verlängerung Kardiologisches Monitoring empfohlen Panobinostat und Makrolide kontraindiziert |
| Panobinostat | Johanniskraut, Rifampicin | Plasmakonzentration von Panobinostat verringert durch CYP3A4-Induktion Kontraindiziert |
4.8 Umgang mit Wechselwirkungen
Bei Verdacht auf Wechselwirkungen in Form von verstärkter Toxizität oder verminderter Wirkung sollte geprüft werden, ob eine Wechselwirkung für die infrage gestellten Arzneimittel literaturbekannt ist. Dazu können die Fachinformationen oder spezielle Datenbanken durch die Apotheke herangezogen werden. Die einzelnen Datenbanken unterscheiden sich in Art, Umfang, Einstufung des Schweregrads und Evidenzniveau der Informationen sowie hinsichtlich ihrer Eignung für die jeweilige Fragestellung. Neben Stockley’s Drug Interactions als Referenzinstrument sind die Datenbanken drugs.com, Lexi-interact, bccancer.bc.ca, Cancer Drug Interactions Checker (www.cancer-druginteractions.org, kostenfrei) hilfreich. Falls die Wechselwirkungen beschrieben sind, sind die Schwere und die Häufigkeit des Vorkommens festzustellen. Leider werden in vielen Datenbanken keine Angaben zum Schweregrad und der klinischen Relevanz einer Wechselwirkung gemacht und es werden keine Hinweise zu den zu ergreifenden Maßnahmen gegeben. Bestätigt sich der Verdacht auf eine klinisch-relevante Wechselwirkung, ist zu überlegen, welche Maßnahme angebracht ist.
Die Maßnahmen bestehen in der Regel
- aus dem engmaschigen Monitoring von Wirkstoffspiegeln oder klinischen Effekten,
- der Dosisanpassung für einen Interaktionspartner,
- dem Absetzen eines der beiden oder sogar beider verursachenden Arzneimittel,
- dem Prüfen, ob eine alternative Arzneimitteltherapie mit einem geringeren Interaktionspotenzial durchgeführt werden kann.
Beispiele für die Vermeidung von CYP3A4-bedingten Wechselwirkungen sind die Anwendung von Pantoprazol anstelle Omeprazol, Fluvastatin anstelle Simvastatin und Echinocandine anstelle Azolantimykotika. In den Tabellen oberhalb sind mögliche Maßnahmen für sehr schwerwiegende und schwerwiegende Wechselwirkungen angegeben. Für die Antitumortherapeutika selbst kommt keine Substitution, sondern nur die Dosisanpassung infrage.
Literatur
Bornmann L, Herdrich K (Hrsg) (2012) Drug Interactions in the therapy of malignant tumors. 6th ed., Baxter Oncology, ISBN 978-3-927105-99-7
Filipowski-Geisselmann K, Gutjahr P (2012) Bedeutsame Arzneimittelwechselwirkungen in der pädiatrischen Onkologie – eine handlungsorientierte Übersicht für die Klinische Praxis. Shaker, Aachen
http://medicine.iupui.edu/clinpharm/ddis/main-table
Krause J, Mildner C, Krämer I (2020) Entwicklung und Implementierung einer Übersichtstabelle zu schwerwiegenden Interaktionen bei (hämato)onkologischen Patienten, Krankenhauspharmazie (41):90–98
Rosentreter J, Engl A-K, Wojnowski L, Kühn M, Wölfel T, Krämer I (2020) Optimierung des Interaktionsmanagements zur Prävention potenzieller schwerwiegender Arzneimittelinteraktionen bei onkologischen Patienten, Best Practice Onkologie, DOI 10.1007/s11654-020-00271-y
Schlichtig K; Dürr P; Dörje F; Fromm MF (2019) Arzneimitteltherapiesicherheit bei neuen oralen Antitumortherapeutika, Dtsch Arztebl 116:775–82, DOI: 10.3238/arztebl.2019.0775
Ritter CA, Höckel M (2019) Vorsicht: Interaktionen! Klinikarzt 48(01/02):36–43, DOI 10.1055/a-0842-3514
[1] Die Festlegung des Differenzierungsgrades erfolgt am histopathologischen Präparat. Das Grading für Weichteilsarkome erfolgt teilweise nach anderen Kriterien, z. B. Malignitätsgrad nach Coindre.
[2] Epigenetik: „Vererbbare Modifikationen, die zu veränderten Genexpressionen führen, ohne die primäre Basensequenz der DNS zu verändern“ [3]; z. B. durch Methylierung oder Acetylierung. In embryonischen Zellen: zeitgerechte Genabschaltung durch epigenetisches „silencing“ (im weiteren Sinne „Stilllegung“).
In differenzierten Zellen: Steuerung der gewebespezifischen Genexpression.
In malignen Zellen: silencing von wachstums-/differenzierungsregulierenden Genen.
[3] Hinweis: Trotz des Namens „Programmed Cell Death Protein“ induziert PD-1 den Tumorzelltod nicht direkt. Mit PD-1 werden Autoimmunreaktionen unterbunden. Es führt beispielsweise zum Zelltod durch Apoptose (programmierter Zelltod) von antigenspezifischen T-Zellen, die in Lymphknoten eingewandert sind.